Um Moleküle mit asymmetrischen Kohlenstoffatomen auf der i-Ebene darzustellen, werden häufig die in 18E1 von E. Fischer vorgeschlagenen Projektionen verwendet.
Betrachten wir das Prinzip ihres Aufbaus am Beispiel eines Bromfluorchlormethan-Moleküls. Ausgangspunkt für die Konstruktion von Fischeoa-Projektionen ist das räumliche Modell des Moleküls bzw. dessen keilförmige Projektion.
Positionieren wir das Molekül so, dass nur das Kohlenstoffatom des Bromfluorchlormethan-Moleküls in der Zeichenebene verbleibt, wie in der Abbildung dargestellt:
Projizieren wir alle Atome auf die Zeichenebene (Br und CL von unten nach oben, da sie sich unter der Zeichenebene befinden, und F und H von oben nach unten). Damit die resultierende Projektion von der Strukturformel abweicht, vereinbaren wir, das asymmetrische Kohlenstoffatom nicht darzustellen. Er impliziert in der Fisher-Projektion am Schnittpunkt der vertikalen und horizontalen Linien:

Wie aus dem obigen Beispiel ersichtlich ist, ist die Fischer-Projektion so aufgebaut, dass die Bindungen eines asymmetrischen Atoms mit Substituenten durch vertikale und horizontale (aber nicht geneigte!) Linien dargestellt werden.
Bei der Verwendung von Fisher-Projektionen ist es wichtig, sich daran zu erinnern, dass die vertikale Linie in ihnen Verbindungen darstellt, die sich von uns wegbewegen, und die horizontale Linie Verbindungen darstellt, die auf uns gerichtet sind. Daraus ergeben sich folgende Regeln für die Verwendung von Fischer-Projektionen:
ES IST VERBOTEN:
1) Sie können die Projektion nicht aus der Zeichenebene entfernen (z. B. „im Licht“, also von der anderen Seite des Blattes betrachten).
2) Sie können die Projektion in der Zeichenebene nicht um 90° und 270° drehen.
3) An einem asymmetrischen Atom können nicht zwei beliebige Substituenten vertauscht werden.
DÜRFEN:
1) Sie können die Projektion in der Ebene der Zeichnung um 180° drehen. Bei dieser Drehung bleiben die vertikalen Linien vertikal und die horizontalen -
horizontal.
2) Es ist möglich, eine gerade Anzahl paarweiser Permutationen von Substituenten an einem asymmetrischen Atom durchzuführen.
3) Es ist möglich, eine zirkuläre Umlagerung von drei Substituenten an einem asymmetrischen Atom durchzuführen. Der vierte Stellvertreter bleibt an seiner Stelle.


Asymmetrisch Das Kohlenstoffatom ist an vier nichtäquivalente Gruppen im Glucosemolekül gebunden; zu diesen Atomen gehören Kohlenstoffatome mit Zahlen von 1 bis 5
Antipoden
eine Substanz, die durch Rotationen der Polarisationsebene des Lichts mit entgegengesetztem Vorzeichen und gleicher Größe gekennzeichnet ist und die Identität aller anderen physikalischen und chemischen Eigenschaften aufweist (mit Ausnahme von Reaktionen mit anderen optisch aktiven Substanzen und physikalischen Eigenschaften in einem chiralen Medium).
Racemat- eine äquimolare Mischung zweier Enantiomere( Enantiomere(altgriechisch ἐνάντιος + μέρος – Gegenteil + Teil, Maß) – ein Paar Stereoisomere, die Spiegelbilder voneinander sind und im Raum nicht kompatibel sind). Racemate besitzen keine optische Aktivität und unterscheiden sich auch in ihren Eigenschaften von den einzelnen Enantiomeren. Sie sind Produkte nichtstereoselektiver Reaktionen
Arten von Retzamat
· Racemisches Konglomerat ist eine mechanische Mischung aus Kristallen zweier Enantiomere im Verhältnis 1:1, wobei jeder Kristall aus Molekülen nur eines Enantiomers besteht.
· Racemische Verbindung(echtes Racemat) besteht aus Kristallen, die jeweils Moleküle beider Enantiomere enthalten und deren Verhältnis 1:1 beträgt. Dieses Enantiomerenverhältnis in racemischen Verbindungen bleibt bis zur Ebene des Kristallgitters erhalten.
· Pseudoracemat ist eine feste Lösung zweier enantiomerer Verbindungen, d. h. es handelt sich um eine homogene ungeordnete Mischung von Enantiomeren im Verhältnis 1:1.
Physikalische Eigenschaften
· Optische Aktivität. Racemate zeigen keine optische Aktivität, das heißt, sie drehen die Polarisationsebene des Lichts nicht. Dieses Phänomen wird durch die Tatsache erklärt, dass die optische Drehung bei Enantiomeren das entgegengesetzte Vorzeichen, aber die gleiche Größe hat. Da es sich bei der Rotation um eine additive Größe handelt, ist sie im Fall eines Racemats aufgrund der Kompensation der Enantiomerenbeiträge gleich Null.
· Kristallform. Da Enantiomere enantiomorphe Kristalle bilden, existieren racemische Konglomerate als zwei Arten von Kristallen, die in ihrer Form Spiegelbilder voneinander sind. Es war diese Tatsache, die es L. Pasteur ermöglichte, Kristalle racemischer Tartrate manuell zu trennen.
· Dichte. Nach der 1895 formulierten Wallachschen Regel haben Kristalle von Racematen eine höhere Dichte als Kristalle der einzelnen Enantiomere. Dies hängt sowohl mit thermodynamischen Faktoren als auch mit der Kinetik der Keimbildung und dem Kristallwachstum der racemischen Verbindung zusammen. Diese Regel wurde durch die Analyse einer kristallographischen Datenbank bestätigt.
· Schmelztemperatur. Bei einem racemischen Konglomerat liegt der Schmelzpunkt immer niedriger als der Schmelzpunkt der einzelnen Enantiomere, wie aus dem Phasendiagramm ersichtlich ist. Beispielsweise schmilzt enantiomerenreines Hexagelicen bei 265–267 °C und das Racemat bei 231–233 °C.
Wenn das Racemat wahr ist, was für die meisten organischen Racemate typisch ist, kann sein Schmelzpunkt entweder höher oder niedriger sein als der Schmelzpunkt der Enantiomere. So liegen im Fall von Dimethyltartrat die Schmelzpunkte des reinen Enantiomers und des Racemats bei 43,3 °C bzw. 86,4 °C. Mandelsäureracemat hingegen schmilzt bei einer niedrigeren Temperatur als die enantiomerenreine Substanz (118,0 °C bzw. 132,8 °C). Die Zugabe eines einzelnen Enantiomers zu einem echten Racemat führt immer zu einem niedrigeren Schmelzpunkt, anders als bei Konglomeraten.
In seltenen Fällen, wenn Racemate die Eigenschaften fester Lösungen aufweisen, schmelzen sie bei der gleichen Temperatur wie die einzelnen Enantiomere (für Kampfer - ≈178 ° C).
· Löslichkeit. Die meisten chiralen Verbindungen zeichnen sich durch unterschiedliche Löslichkeiten zwischen dem Racemat und den einzelnen Enantiomeren aus. Die Löslichkeit racemischer Konglomerate ist höher als die Löslichkeit reiner Enantiomere. Meyerhoffers Faustregel, die auf nicht dissoziierende organische Verbindungen anwendbar ist, besagt, dass die Löslichkeit des Racemats doppelt so hoch ist wie die der Enantiomere. Bei echten Racematen kann die Löslichkeit größer oder kleiner sein als die Löslichkeit der Enantiomere


Reaktionen von Monosacchariden









Glucose, oder Traubenzucker, oder Traubenzucker(D-Glukose), C 6 H 12 O 6 – kommt im Saft vieler Früchte und Beeren vor, darunter auch Weintrauben, daher der Name dieser Zuckerart. Es ist ein Monosaccharid und ein Sechs-Hydroxy-Zucker (Hexose). Die Glukoseeinheit ist Bestandteil von Polysacchariden (Zellulose, Stärke, Glykogen) und einer Reihe von Disacchariden (Maltose, Laktose und Saccharose), die im Verdauungstrakt beispielsweise schnell in Glukose und Fruktose zerlegt werden.


D-Fruktose

Erhalten als β-Form. Sehr hygroskopische farblose Prismen oder Nadeln. tschmelzen 103-105 (zerfällt).
Spezifische optische Drehung für die Natrium-D-Linie bei einer Temperatur von 20 °C: [α] D 20 -132,2 → -92,4 (c=4 in H 2 O).
Löslichkeit: 375 20, 740 55 in H 2 O; löslich in MeOH, EtOH, Pyridin, Aceton, Eisessig.
Die wasserfreie Form ist bei Temperaturen > 21,4 °C stabil. Kann bei Temperaturen hydratisiert werden, um Hemihydrat (und Dihydrat) zu bilden< 20°С. Перекристаллизовать из МеОН. Положительная реакция Селиванова. Кристаллический сахар - β-D-пираноза, но в растворе содержится ≥ 15% фуранозной формы и значительное количество открытой линейной формы. В составе соединений найдена только фуранозная форма. Сладкий вкус.
Askorbinsäure(aus dem Altgriechischen ἀ – nicht- + lat. Skorbutus- Skorbut) - eine organische Verbindung mit der Formel C 6 H 8 Ö 6, ist einer der Hauptstoffe in der menschlichen Ernährung, der für die normale Funktion des Binde- und Knochengewebes notwendig ist. Erfüllt die biologischen Funktionen eines Reduktionsmittels und Coenzyms einiger Stoffwechselprozesse und ist ein Antioxidans. Nur eines der Isomere ist biologisch aktiv – L- Ascorbinsäure, die genannt wird Vitamin C . Ascorbinsäure kommt natürlicherweise in vielen Obst- und Gemüsesorten vor.

Glykoside- organische Verbindungen, deren Moleküle aus zwei Teilen bestehen: einem Kohlenhydratrest (Pyranosid oder Furanosid) und einem Nichtkohlenhydratfragment (dem sogenannten Aglycon). Im allgemeineren Sinne können auch Kohlenhydrate, die aus zwei oder mehr Monosaccharidresten bestehen, als Glykoside betrachtet werden. Meist kristalline, seltener amorphe Stoffe, gut löslich in Wasser und Alkohol.
Glykoside sind eine große Gruppe organischer Substanzen, die in der Pflanzenwelt (seltener in der Tierwelt) vorkommen und/oder synthetisch gewonnen werden. Bei der sauren, alkalischen oder enzymatischen Hydrolyse werden sie in zwei oder mehr Komponenten gespalten – ein Aglycon und ein Kohlenhydrat (oder mehrere Kohlenhydrate). Viele der Glykoside sind toxisch oder haben starke physiologische Wirkungen, beispielsweise Glykoside von Digitalis, Strophanthus und anderen.

Fruktose(Fruchtzucker), C 6 H 12 O 6 - Monosaccharid, Ketonalkohol, Ketohexose, Glucoseisomer.

Physikalische Eigenschaften
Weiße kristalline Substanz, gut wasserlöslich. Der Schmelzpunkt von Fructose ist niedriger als der Schmelzpunkt von Glucose. 2-mal süßer als Glukose und 4-5-mal süßer als Laktose.
Chemische Eigenschaften
In wässrigen Lösungen liegt Fructose als Tautomerenmischung vor, in der β-D-Fructopyranose vorherrscht und bei 20 °C etwa 20 % β-D-Fructofuranose und etwa 5 % α-D-Fructofuranose enthält


Im Gegensatz zu Glucose und anderen Aldosen ist Fructose sowohl in alkalischen als auch in sauren Lösungen instabil; zersetzt sich unter den Bedingungen der sauren Hydrolyse von Polysacchariden oder Glykosiden
Diese Formeln werden üblicherweise zur räumlichen Darstellung enthaltender Moleküle verwendet asymmetrisches Kohlenstoffatom : das ist tetraedrisch ( sp 3-Hybrid-Kohlenstoffatom, das an vier verschiedene Atome oder Atomgruppen gebunden ist.
Zur Darstellung der Projektionsformel wird das tetraedrische Modell des Moleküls so positioniert, dass sich das asymmetrische Atom in der Zeichenebene befindet und zwei Bindungen des asymmetrischen Kohlenstoffatoms in einer horizontalen Ebene liegen, die aus der Zeichenebene zum Betrachter herausragt . Die anderen beiden Anschlüsse sollten in einer vertikalen Ebene liegen, die sich vom Betrachter über die Zeichenebene hinaus erstreckt.
Bei der Verwendung von Fischer-Projektionen wird das Symbol für das asymmetrische Kohlenstoffatom normalerweise weggelassen:
Es sollte immer berücksichtigt werden, dass Fishers Formeln Projektionen auf eine Ebene sind und nicht als räumliche Modelle betrachtet werden sollten.
Daher sind folgende Regeln zu beachten:
die einem bestimmten Stereoisomer entsprechende Projektionsformel kann nicht aus der Zeichenebene entfernt und in der Ebene nicht um 90 ° und 270 ° gedreht werden;
Die Formel kann um 180° gedreht werden

Wenn sich die Position einer Atomgruppe nicht ändert, können die anderen drei im oder gegen den Uhrzeigersinn gedreht werden:

4.2. Optische Aktivität* und Chiralität
Optische Aktivität bezieht sich auf die Eigenschaft, die Fähigkeit einer Substanz, die Ebene des polarisierten Lichts zu drehen (um einen bestimmten Winkel abzuweichen).
Die Drehung der Polarisationsebene erfolgt entweder im Uhrzeigersinn (Rechtsdrehung) oder gegen den Uhrzeigersinn (Linksdrehung).
Für eine Verbindung, die ein asymmetrisches Kohlenstoffatom enthält, sind zwei Isomere möglich, die als Objekt und als Spiegelbild miteinander in Beziehung stehen, wie am Beispiel der Milchsäure zu sehen ist.


Man nennt zwei Isomere, die jeweils ein Spiegelbild des anderen sind optische oder Spiegelisomere .
Ein solches Paar von Spiegelisomeren heißt Enantiomere oder optische Antipoden .
Enantiomere haben bis auf die Drehrichtung der Polarisationsebene die gleichen physikalischen Eigenschaften: Sie lenken polarisiertes Licht im gleichen Winkel, aber in entgegengesetzte Richtungen ab.
Verbindungen, die mit ihrem Spiegelbild nicht kompatibel sind, werden genannt chiral .
Chiralität (von cheir – Hand) ist ein Grundkonzept der Stereochemie und bezeichnet die Eigenschaft eines Objekts, mit seinem Spiegelbild inkompatibel zu sein; ist eine Voraussetzung für die optische Aktivität von Molekülen.
Man bezeichnet ein Stoffgemisch, das aus gleichen Mengen beider Enantiomere besteht racemische Mischung . Die optische Aktivität einer solchen Mischung beträgt 0.
Man nennt Stereoisomere, die keine Enantiomere sind Diastereomere (Einzelheiten siehe unten, Abschnitt 4.4.).
Somit sind alle chiralen Moleküle Moleküle optisch aktiver Verbindungen.
Es besteht eine Eins-zu-eins-Korrespondenz zwischen Chiralität und optischer Aktivität. Ein charakteristisches Merkmal chiraler Moleküle ist das Fehlen einer Achse, eines Zentrums und einer Symmetrieebene Elemente der Symmetrie .
Symmetrieachse. Bringt die Drehung um eine Achse, die unter einem Winkel von 2π/n=360 o/n durch das Molekül verläuft, dieses wieder in seinen ursprünglichen Zustand, so nennt man eine solche Achse Symmetrieachse n-ter Ordnung С n . Es ist klar, dass beim Wert n=1 in jedem Fall die Drehung um die Achse das Molekül in seinen ursprünglichen Zustand zurückführt.
Beispielsweise hat ein Wassermolekül (a) eine Symmetrieachse zweiter Ordnung (n=2), Chlormethan (b) eine dritte Ordnung und in Benzol (c) zusammen mit sechs Achsen, die in der Kreisebene liegen (n =2) gibt es eine weitere Achse (n=6), die senkrecht zur Kreisebene steht.

Symmetrieebene. Wenn eine durch ein Molekül (Objekt) verlaufende Ebene es in zwei Teile teilt, die als Spiegelisomere zueinander in Beziehung stehen, spricht man von einem solchen Symmetrieebene . Wasser hat zwei Symmetrieebenen und Chlormethan drei.

Symmetriezentrum i- Dies ist ein Punkt in der Mitte eines Moleküls, in gleichem Abstand, von dem zwei äquivalente, äquivalente Punkte eines bestimmten Moleküls auf derselben Geraden liegen.
Ein Molekül kann nicht mehr als ein Symmetriezentrum haben.
Unter dem Gesichtspunkt der Anwesenheit oder Abwesenheit von Symmetrieelementen in Molekülen ist es daher üblich, darüber zu sprechen Chiralität Und Achiralität . In den meisten Fällen stimmt ihre Bedeutung mit den Begriffen „Asymmetrie“ und „Symmetrie“ überein. Allerdings ist Chiralität ein viel weiter gefasster Begriff als Asymmetrie, da ein chirales Molekül einige Symmetrieelemente enthalten kann. Aus dem gleichen Grund wird in der Stereochemie der Begriff „asymmetrisches“ Atom sehr oft durch den Begriff „Chiralitätszentrum“ ersetzt. Daher sollte Chiralität als die Eigenschaft eines Objekts verstanden werden, mit seinem Spiegelbild inkompatibel zu sein. Ein achirales Molekül enthält mindestens ein Symmetrieelement (entweder ein Zentrum oder eine Symmetrieebene). Ein Sonderfall der Achiralität ist Chiralität: die Fähigkeit, Eigenschaft eines achiralen Moleküls, sich durch eine einmalige Änderung in einem Strukturfragment in ein chirales umzuwandeln. In der Regel ist eine solche Operation möglich, wenn dies der Fall ist prochirales Zentrum – also ein Zentrum, das zwei verschiedene und zwei identische Substituenten enthält. Durch Ersetzen eines dieser identischen Substituenten durch einen anderen, der sich von allen vorhandenen unterscheidet, wird dieses Zentrum zu einem chiralen:

Konfiguration und Konformation
Stereoisomere können variieren Konfigurationen (Konfigurationsisomere) oder Konformationen (Konformationsisomere).
Aufbau– die räumliche Anordnung von Atomen und/oder Atomgruppen um das chirale Zentrum, die Doppelbindungsebene oder die Ringebene und charakterisiert ein bestimmtes Stereoisomer. Dieses Konzept ist größtenteils qualitativer Natur und spiegelt im Wesentlichen das stereochemische Merkmal der spezifischen Anordnung von Atomen im Raum um das Chiralitätszentrum eines bestimmten Moleküls wider.
Nachfolgend sind Verbindungspaare aufgeführt, bei denen es sich um Konfigurationsisomere handelt:

Konformation*- eine bestimmte Geometrie des Moleküls aufgrund der inneren Rotation von Atomen oder Atomgruppen um einfache Bindungen. In diesem Fall bleibt die stereochemische Konfiguration des Moleküls unverändert.
Zur Darstellung von Konformationen werden Newman-Projektionen verwendet. Für Ethan sind zwei Grenzkonformationen möglich: verfinstert (I) und inhibiert (II).

Butan unterscheidet sich von Ethan durch das Vorhandensein von zwei Methylgruppen, weshalb für dieses Molekül eine größere Anzahl inhibierter Konformationen möglich ist, wie unten gezeigt. Die stabilere Konformation von I wird als bezeichnet Anti-: Hier sind die Methylgruppen möglichst weit voneinander entfernt (Diederwinkel beträgt 180°). Die anderen beiden Konformationen sind schräge oder gauche-Konformationen (II und III), in denen die Methylgruppen einen Winkel von 60° bilden:

Bei der Darstellung von Stereoisomeren werden häufig Fischersche Formeln verwendet. In diesen Formeln wird das Chiralitätszentrum mit vier Bindungen dargestellt, die im rechten Winkel zueinander stehen. Die vertikalen Linien stellen die Projektion der Substituenten dar, die sich hinter der Ebene befinden, während die horizontalen Linien die Projektion der Substituenten darstellen, die sich vor der Ebene befinden. In Fischers Projektionsformeln wird üblicherweise das Symbol für das asymmetrische Kohlenstoffatom weggelassen.
Vor 1951 war es unmöglich, eine absolute Konfiguration festzulegen. Rozanov schlug 1906 die Verwendung von rechtsdrehendem (+) Glycerinaldehyd als relativen Standard vor, dem willkürlich die Konfiguration D zugeordnet wurde. Der linksdrehende Antipode wurde mit dem Buchstaben L bezeichnet.
D-Glycerinaldehyd L-Glycerinaldehyd
In Fischers Formeln wird die längste Kohlenstoffkette vertikal geschrieben, mit Kohlenstoffatom Nr. 1 oben; Die vertikalen Bindungen des asymmetrischen Kohlenstoffatoms liegen hinter der Zeichenebene, die horizontalen oberhalb der Zeichenebene.

D-Milchsäure L-Milchsäure
Wenn wir in der Fischer-Projektion zwei benachbarte Gruppen vertauschen, erhalten wir ein Spiegelbild der ursprünglichen Verbindung. Ein Spiegelbild der Ausgangsstruktur erhält man auch durch Drehung der Fischer-Projektion um 90 Grad.
Diastereoisomerie
Verbindungen, deren Moleküle zwei oder mehr Stereozentren aufweisen, können als Diastereomere vorliegen. Mit zunehmender Anzahl asymmetrischer Kohlenstoffatome nimmt die Anzahl der Stereoisomere mit dem Auftreten jedes neuen Stereozentrums zu und kann nach der Formel N = 2n berechnet werden, wobei N– Anzahl der Stereozentren. Moleküle mit zwei asymmetrischen Kohlenstoffatomen können als vier Stereoisomere existieren. Beispielsweise hat das 2,3-Dibrompentan-Molekül zwei Stereozentren und daher hat diese Verbindung 4 Stereoisomere.
![]()
2,3-Dibrompentan
(2S,3R)-2,3-Dibrompentan (2R,3S)-2,3-di... (2S,3S)-2,3-di... (23,3R)-2,3-di ...
(I) (II) (III) (IV)




Paare von Stereoisomeren (I) und (II), auch (III) und (IV) beziehen sich aufeinander als ein Objekt und ein inkompatibles Spiegelbild, d. h. sind Enantiomerenpaare. Stereoisomere in allen anderen Paaren sind Diastereomere. Zwei unterschiedliche Konfigurationen desselben Moleküls, die keine Enantiomere sind, werden Diastereomere genannt. Die beiden Diastereomere unterscheiden sich in allen Eigenschaften und sind als zwei verschiedene Verbindungen relativ einfach zu trennen.
In den Projektionsformeln (I) und (II) befinden sich identische Liganden auf derselben Seite der Projektion; solche Stereoisomere nennt man erythro-Formen. In den Formeln (III) und (IV) befinden sich die gleichen Liganden auf gegenüberliegenden Seiten der vertikalen Linie der Fischer-Projektion; die entsprechenden Verbindungen werden aufgerufen Trio-Formen.
A. Mesoverbindungen
Eine Struktur mit zwei Stereozentren weist möglicherweise nicht immer 4 Stereoisomere auf. Beispielsweise hat 2,3-Dibrombutan zwei Stereozentren, aber nicht 4, sondern nur 3 Stereoisomere.
(2S,3R)-2,3-Dibrombutan (2S,3S)-2,3-di... (2R,3R)-2,3-di...
Meso- bilden
Die Atome von 2,3-Dibrombutan können von oben nach unten oder von unten nach oben nummeriert werden, dann ist klar, dass die ersten beiden Strukturen dasselbe Stereoisomer darstellen. Dieses Stereoisomer ist achiral und optisch inaktiv, da es eine Symmetrieebene aufweist

Ex. 7. Zeichnen Sie die Fischer-Formeln der räumlichen Isomere: (a) Glycerinaldehyd (2,3-Dihydroxypropanal), (b) Milchsäure (2-Hydroxypropansäure), (c) Äpfelsäure (2-Hydroxybutandisäure oder Hydroxybernsteinsäure), (d) Weinsäure (2). ,3-Dihydroxybutandisäure oder Dihydroxybernsteinsäure.
P-Diastereomere
Alkene und ihre Derivate mit der allgemeinen Formel ABC=CDE können in der Form vorliegen p-Diastereomere. p-Diastereomere entstehen, wenn die mit einzelnen Kohlenstoffatomen der Doppelbindung verbundenen Liganden nicht identisch sind. p-Diastereomere unterscheiden sich voneinander durch unterschiedliche Positionen der Liganden relativ zur Symmetrie der p-Bindung.
Substituenten, die sich auf einer Seite der Doppelbindung befinden, nennt man in cis-Position relativ zueinander; Wenn sie sich auf gegenüberliegenden Seiten der Doppelbindungsebene befinden, dann ist dies der Fall Trance-Position. Neuerdings statt Begriffe cis- Und Trance- Z,E-System wird empfohlen. Befinden sich die beiden höchsten Gruppen (nach dem Cahn-Ingold-Prelog-System) auf derselben Seite der p-Bindung, so wird die Konfiguration der Substituenten mit dem Symbol Z bezeichnet, liegen diese Gruppen jedoch auf gegenüberliegenden Seiten von die p-Bindungsebene, dann wird die Konfiguration mit dem Symbol E bezeichnet.

Daher haben wir zwei Arten der Diastereoisomerie diskutiert:
Diastereoisomerie, die aus einer Kombination chiraler Elemente resultiert (in diesem Fall überlagern sich Diastereoisomerie und Enantiomerie);
Diastereoisomerie cis-trans-Isomere.
Ex. 8.. Schreiben Sie die Strukturformel(en) cis- 1,2-Dichlorethen und Trance-1,2-Dichlorethen, (b) cis- 1,2-Difluorethen und Trance-1,2-Difluorethen, (c) cis- 1,2-Dichlor -
1,2-Difluorethen und Trance- 1,2-Dichlor-1,2-difluorethen.
2.4 Cis-trans-Isomerie und Konformationen von Cycloalkanen
Cyclopropan, ein Mittel zur Inhalationsanästhesie (Siedepunkt -33 °C), hat eine flache Struktur. Jedes der drei Wasserstoffatome nimmt eine Seite der Ringebene ein Trance Position relativ zu jedem Wasserstoffatom, das sich auf der anderen Seite der Ringebene befindet. Zwei beliebige Wasserstoffatome, die sich auf derselben Seite des Rings befinden, sind in cis positionieren und verdecken sich gegenseitig.

Cyclopropan cis-Wasserstoffe Trance-Wasserstoffe
Es gibt nur ein monosubstituiertes Cyclopropan. Disubstituiertes Cyclopropan mit identischen Substituenten kann in Form von zwei Diastereomeren vorliegen. Traditionell werden sie als beschrieben cis- Und Trance- Formen. U cis-Form ist eine Symmetrieebene und kann daher nicht als Enantiomerenpaar existieren Trance-form - vielleicht.

 cis-1,2-Dimethylcyclopropan Trance-1,2-Dimethylcyclopropan
cis-1,2-Dimethylcyclopropan Trance-1,2-Dimethylcyclopropan
B. Cyclopentan
Das Cyclopentanmolekül ist nahezu flach. Das 1,2-Dimethylcyclopentan-Molekül besitzt zwei Stereozentren und liegt daher in Form von drei Stereoisomeren vor.
Enantiomere Meso-Verbindung
Das trans-Isomer liegt in zwei Enantiomeren vor und cis-1,2-Dimethylcyclopentan ist meso Verbindung, weil es eine Symmetrieebene hat. 1,3-Dimethylcyclopentan existiert auch in Form von drei Stereoisomeren.


Enantiomere Meso-Verbindung
B. Cyclohexan
Wenn der Cyclohexanring mehr als einen Substituenten enthält, werden bei der Beurteilung der Stabilität einer bestimmten Konformation die relative Position der Substituenten im Ring und ihre Struktur berücksichtigt. Also in einem Molekül Trance-1,2-Dimethylcyclohexan, beide Substituenten können entweder eine axiale oder äquatoriale Position einnehmen; Vorteilhafter ist natürlich die diquatoriale Konformation.


Trance-1,2-Dimethylcyclohexan cis-1,2-Dimethylcyclohexan
U cis-Isomer in einer der beiden Sesselkonformationen, wobei eine der Methylgruppen eine axiale Position einnimmt, die andere eine äquatoriale Position.
Das Cyclohexanmolekül kann mehrere Konformationen annehmen.

„Sessel“ „Badewanne“
Bei normalen Temperaturen liegen 99,9 % davon in Form zweier sich schnell ineinander umwandelnder Sesselkonformationen vor. Die Sesselkonformation ist am symmetrischsten, wobei jedes Kohlenstoffatom zwei nichtäquivalente CH-Bindungen aufweist.

Die Form des Stuhls weist eine Symmetrieachse dritter Ordnung auf. Die sechs C-H-Bindungen in Cyclohexan verlaufen parallel zu dieser Achse: drei davon sind nach oben und drei nach unten gerichtet, diese Wasserstoffatome nehmen eine axiale (a) Position ein. Weitere sechs C-H-Bindungen stehen nahezu senkrecht zur Symmetrieachse; diese Wasserstoffatome nehmen eine äquatoriale (e) Position ein.
Bei Konformationsumwandlungen sind alle axialen Substituenten A werden äquatorial und dementsprechend äquatorial e– axial. Unter diesem Gesichtspunkt werden Konformationsübergänge von Cyclohexan genannt Umkehrung.

Der Übergang erfolgt durch eine dazwischenliegende Twist-Konformation. Die Barriere für den Konformationsübergang in Cyclohexan beträgt etwa 42 kJ/mol und ändert sich kaum mit der Einführung von Substituenten.
Aus der Newman-Stuhlprojektion von Cyclohexan geht hervor, dass benachbarte Wasserstoffatome nicht verdeckt werden.


Newman-Projektion von Cyclohexan
Neben der Stuhlform gibt es auch eine Badewannenform, eine Halbstuhl- (oder Halbdreh-)Form und eine Twist-Form aus Cyclohexan. Diese Formen werden als bewegliche Formen bezeichnet.

Abb.4. Energieeigenschaften von Cyclohexan-Konformationen
In der Badewannenform werden zwei Wasserstoffatome als Buschabdruck und zwei als Fahnenmast bezeichnet. Die Badewannenform ist ein Übergang zwischen verschiedenen Twist-Formen und die Half-Twist-Form ist ein Übergang zwischen der Stuhlform und der Twist-Form. Von den drei beweglichen Formen spielt die Twist-Form die wichtigste Rolle.
Darüber hinaus findet durch die Half-Twist-Konformation ein wichtiger Prozess der gegenseitigen Stuhl-Stuhl-Umwandlung (Inversion) statt, wodurch alle (a) Bindungen zu (e) werden (gleichzeitig alle cis-trans-Beziehungen zwischen sie bleiben unverändert).
Für monosubstituierte Cyclohexane gibt es zwei nichtäquivalente Konformationen mit Substituenten in axialer und äquatorialer Position. Normalerweise ist die Äquatorform stabiler.

Der Unterschied in der relativen Stabilität von Konformeren mit axialer und äquatorialer Position der Substituenten wird erklärt durch diaxiale Wechselwirkung axiale Wasserstoffatome mit Substituenten.



Newman-Projektionen von Methylcyclohexan-Konformeren
Die Barriere für die gegenseitige Umwandlung von (a)- und (e)-Konformeren ist sehr gering. Die meisten Substituenten bevorzugen die äquatoriale Position. reibt-Die Butylgruppe in Cyclohexan ist praktisch immer äquatorial.
Moderne Vorstellungen über die Struktur organischer Verbindungen. Grundlagen der Stereochemie organischer Verbindungen. Asymmetrisches Kohlenstoffatom. Chiralität. Fischer-Projektionsformeln.
Theorie der chemischen Struktur A.M. Butlerow
Im Jahr 1861 A.M. Butlerov schlug eine Theorie der chemischen Struktur organischer Verbindungen vor, die aus den folgenden Grundprinzipien besteht.
1) In den Stoffmolekülen gibt es eine strenge Abfolge der chemischen Bindung von Atomen, die als chemische Struktur bezeichnet wird.
2) Die chemischen Eigenschaften eines Stoffes werden durch die Art seiner Elementarbestandteile, deren Menge und chemische Struktur bestimmt.
3) Wenn Stoffe mit gleicher Zusammensetzung und gleichem Molekulargewicht unterschiedliche Strukturen aufweisen, kommt es zum Phänomen der Isomerie.
4) Da sich bei bestimmten Reaktionen nur einige Teile des Moleküls verändern, hilft die Untersuchung der Struktur des Produkts dabei, die Struktur des ursprünglichen Moleküls zu bestimmen.
5) Die chemische Natur (Reaktivität) einzelner Atome in einem Molekül ändert sich je nach Umgebung, d. h. abhängig davon, mit welchen Atomen anderer Elemente sie verbunden sind.
Butlerovs Theorie bietet die grundlegende Möglichkeit, die Geometrie eines Moleküls (mikroskopische Eigenschaften) durch Kenntnis der chemischen Eigenschaften (makroskopische Eigenschaften) zu kennen. Die Grundprinzipien der Strukturtheorie behalten bis heute ihre Bedeutung.
Elektronische Theorien der chemischen Bindung.
Die elektronische Struktur organischer Verbindungen wird mithilfe von Lewis-Elektronenformeln dargestellt. Sie verwenden Punkte, um die Position aller Valenzelektronen anzuzeigen: Elektronen chemischer Bindungen und freie Elektronenpaare. Es wird angenommen, dass einzelne Elektronenpaare Teil der Außenhülle nur eines Atoms sind und die an der Bildung einer kovalenten Bindung beteiligten Elektronen Teil der Außenhülle beider Atome sind. In der Lewis-Formel für Kohlenstofftetrachlorid unten haben beispielsweise alle Atome ein Oktett von Elektronen.
Für jedes Atom in einer Lewis-Struktur wird eine formale Ladung bestimmt. Es wird angenommen, dass das Atom alle ungeteilten Elektronen und die Hälfte der Elektronen in kovalenten Bindungen besitzt. Ein Überschuss an Elektronen eines Atoms in einem Molekül im Vergleich zu einem freien Atom verursacht eine negative Ladung, und ein Mangel verursacht eine positive Ladung. Die Summe der formalen Ladungen aller Atome ergibt die Ladung des Teilchens als Ganzes.

Grundprinzipien der quantenorganischen Chemie.

Moderne Theorien kovalenter Bindungen basieren auf den Konzepten der Quantenmechanik. Nach den Prinzipien der Quantenmechanik wird der Zustand eines Elektrons in einem Atom durch eine Wellenfunktion bestimmt, die Atomorbital genannt wird. Die Bildung einer chemischen Bindung zwischen Atomen wird als Ergebnis der Wechselwirkung zweier Orbitale betrachtet, die jeweils ein Elektron enthalten. In diesem Fall kommt es zur Bildung von Molekülorbitalen (MOs). Aus zwei Atomorbitalen entstehen zwei Molekülorbitale, von denen eines ( verbinden) hat eine geringere Energie und der andere ( Lockerung) – höhere Energie als das ursprüngliche AO.
Die Bindungselektronen besetzen das Bindungsorbital mit der niedrigeren Energie, sodass die Wechselwirkung der Orbitale zu einem Energiegewinn führt.

Abhängig von der Art der Atomorbitale, die sich verbinden, werden unterschiedliche Arten von MOs gebildet. Dabei spielen die Symmetrie und Knoteneigenschaften von Orbitalen eine entscheidende Rolle. Atomar S -Orbitale haben sphärische Symmetrie und keine Knotenflächen, die durch die Mitte des Atoms verlaufen. Atomar P -Orbitale haben Zylindersymmetrie und drei Zustände p x , p y und p z . Jeden p -Orbital hat eine Knotenebene, die durch den Mittelpunkt des Atoms bzw. senkrecht zur Achse verläuft x, y oder z.
Die Knotenoberfläche ist der Ort, an dem die Wahrscheinlichkeit, ein Elektron zu finden, Null ist und die Wellenfunktion das Vorzeichen ändert. Je mehr Knoten vorhanden sind, desto höher ist die Energie des Orbitals. Auf diese Weise, P -Orbital besteht aus zwei Teilen, in denen die Vorzeichen der Wellenfunktionen entgegengesetzt sind.
Bei der Betrachtung der elektronischen Struktur mehratomiger Moleküle ist es notwendig, einen Satz von Orbitalen zu verwenden, der ihre maximale Überlappung erreicht. Diesbezüglich gibt es ein Konzept Hybridisierung Orbitale. Ein Kohlenstoffatom im angeregten Zustand enthält in seinem äußeren Energieniveau vier ungepaarte Elektronen und ist in der Lage, vier kovalente Bindungen zu bilden.
Hybridorbitale sind an der Bindungsbildung beteiligt.
Der erste Valenzzustand ist die sp3-Hybridisierung . Durch die Hybridisierung unter Beteiligung eines s- und drei p-Orbitals des Kohlenstoffatoms entstehen vier äquivalente sp 3 -Hybridorbitale, die in Winkeln von 109,5 o auf die Eckpunkte des Tetraeders gerichtet sind:
Im Zustand der sp 3 -Hybridisierung bildet das Kohlenstoffatom vierS-Bindungen mit vier Substituenten und hat eine tetraedrische Konfiguration mit Bindungswinkeln gleich oder nahe 109,5 °:

Methan
Der zweite Valenzzustand ist die sp 2 -Hybridisierung . Durch die Hybridisierung unter Beteiligung eines s- und zweier p-Orbitals eines Kohlenstoffatoms entstehen drei äquivalente sp 2 -Hybridorbitale, die in einem Winkel von 120 ° in derselben Ebene liegen und an denen das p-Orbital nicht beteiligt ist bei der Hybridisierung liegt senkrecht zur Ebene der Hybridorbitale.
Im Zustand der sp 2 -Hybridisierung bildet das Kohlenstoffatom dreiS-Bindungen aufgrund von Hybridorbitalen und einemP-Bindung, da das p-Orbital nicht an der Hybridisierung beteiligt ist und drei Substituenten aufweist.
Der dritte Wertigkeitszustand von Kohlenstoff ist die sp-Hybridisierung . Durch die Hybridisierung unter Beteiligung eines s- und eines p-Orbitals entstehen zwei äquivalente sp-Hybridorbitale, die in einem Winkel von 180 0 liegen, und die nicht an der Hybridisierung beteiligten p-Orbitale liegen senkrecht zur Ebene der Hybridorbitale und untereinander. Im sp-Hybridisierungszustand bildet das Kohlenstoffatom zweiS-Bindungen aufgrund von Hybridorbitalen und zweiP-Bindung aufgrund von p-Orbitalen, die nicht an der Hybridisierung teilnehmen, und hat zwei Substituenten:

Acetylen
Grundlagen der Stereochemie.
Die Stereochemie ist ein Teil der Chemie, der sich mit der Untersuchung der räumlichen Struktur von Molekülen und dem Einfluss dieser Struktur auf die physikalischen und chemischen Eigenschaften einer Substanz sowie auf die Richtung und Geschwindigkeit ihrer Reaktionen befasst.
Konformationen (Rotationsisomerie).
Der Übergang vom einfachsten organischen Kohlenwasserstoff, Methan, zu seinem nächsten Homologen, Ethan, wirft Probleme der räumlichen Struktur auf, zu deren Lösung es nicht ausreicht, die zuvor diskutierten Parameter zu kennen. Ohne Änderung der Bindungswinkel oder Bindungslängen kann man sich viele geometrische Formen des Ethanmoleküls vorstellen, die sich voneinander durch die gegenseitige Drehung der Kohlenstofftetraeder um die sie verbindende C-C-Bindung unterscheiden. Als Ergebnis dieser Rotation Rotationsisomere (Konformere) . Die Energie verschiedener Konformere ist nicht gleich, aber die Energiebarriere, die die verschiedenen Rotationsisomere trennt, ist bei den meisten organischen Verbindungen gering. Daher ist es unter normalen Bedingungen in der Regel unmöglich, Moleküle in einer streng definierten Konformation zu fixieren. Typischerweise existieren mehrere Rotationsformen, die sich leicht ineinander umwandeln, im Gleichgewicht nebeneinander.
Betrachten wir Möglichkeiten zur grafischen Darstellung von Konformationen und deren Nomenklatur. Für ein Ethanmolekül kann man die Existenz zweier Konformationen vorhersagen, die sich energetisch maximal unterscheiden. Sie werden unten als dargestellt Perspektivische Projektionen (1) („Sägeziegen“), seitliche Vorsprünge (2 und Newmans Formeln .

Die links dargestellte Konformation wird aufgerufen verdeckt . Dieser Name erinnert uns daran, dass die Wasserstoffatome beider CH 3 -Gruppen einander gegenüberstehen. Die verfinsterte Konformation hat eine erhöhte innere Energie und ist daher ungünstig. Die rechts dargestellte Konformation wird aufgerufen gehemmt , was bedeutet, dass die freie Rotation um die C-C-Bindung in dieser Position „gehemmt“ ist, d. h. das Molekül liegt überwiegend in dieser Konformation vor.
Je komplexer das Molekül wird, desto mehr mögliche Konformationen gibt es. Ja für N-Butan kann bereits in sechs Konformationen dargestellt werden, die sich in der relativen Anordnung der CH 3 -Gruppen unterscheiden, d. h. Drehung um die zentrale Verbindung C-C. Nachfolgend sind die Konformationen von n-Butan als Newman-Projektionen dargestellt. Die links dargestellten Konformationen (schattiert) sind energetisch ungünstig; nur gehemmte werden praktisch realisiert.

Die verschiedenen verfinsterten und gehemmten Konformationen von Butan sind energetisch nicht gleich. Entsprechende Energien aller Konformationen, die während der Rotation um die zentrale C-C-Bindung entstehen.
Konformationen sind also verschiedene räumliche Formen eines Moleküls, das eine bestimmte Konfiguration aufweist. Konformere sind stereoisomere Strukturen, die Energieminima im Potentialenergiediagramm entsprechen, sich im mobilen Gleichgewicht befinden und durch Rotation um einfache Bindungen zur gegenseitigen Umwandlung fähig sind.
Manchmal wird die Barriere für solche Umwandlungen hoch genug, um stereoisomere Formen (z. B. optisch aktive Biphenyle) zu trennen. In solchen Fällen spricht man nicht mehr von Konformeren, sondern von tatsächlich existierenden Stereoisomere .
Geometrische Isomerie.
Eine wichtige Konsequenz der Starrheit einer Doppelbindung (das Fehlen einer Rotation um sie herum) ist ihre Existenz geometrische Isomere . Die häufigsten davon sind cis-, trans-Isomere Verbindungen der Ethylenreihe mit ungleichen Substituenten an ungesättigten Atomen. Das einfachste Beispiel sind die Isomere von Buten-2.

Geometrische Isomere haben die gleiche chemische Struktur, unterscheiden sich jedoch in der räumlichen Anordnung der Atome, d.h. Von Konfigurationen . Dieser Unterschied führt zu einem Unterschied in den physikalischen (sowie chemischen) Eigenschaften. Geometrische Isomere können im Gegensatz zu Konformeren in reiner Form isoliert werden und liegen als einzelne stabile Substanzen vor. Für ihre gegenseitige Umwandlung ist eine Energie in der Größenordnung von 125–170 kJ/mol erforderlich, die durch Erhitzen oder Bestrahlen bereitgestellt werden kann.
Im einfachsten Fall bereitet die Nomenklatur geometrischer Isomere keine Schwierigkeiten: cis- Formen sind geometrische Isomere, bei denen identische Substituenten auf derselben Seite der Pi-Bindungsebene liegen. Trance- Isomere haben identische Substituenten auf verschiedenen Seiten der Pi-Bindungsebene. In komplexeren Fällen wird es verwendet Z,E-Nomenklatur . Sein Hauptprinzip: die Konfiguration angeben cis-(Z, aus dem Deutschen Zusammen – zusammen) oder Trance-(E, vom Deutschen Entgegen – gegenüber) Standort hochrangige Stellvertreter mit einer Doppelbindung.
Im Z,E-System gelten Substituenten mit einer höheren Ordnungszahl als höher. Wenn die Atome, die direkt mit ungesättigten Kohlenstoffen verbunden sind, gleich sind, wandern sie bei Bedarf in die „zweite Schicht“ – in die „dritte Schicht“ usw.
3. Optische Isomerie (Enantiomerie).
Unter den organischen Verbindungen gibt es Stoffe, die die Polarisationsebene des Lichts drehen können. Dieses Phänomen nennt man optische Aktivität, und die entsprechenden Stoffe heißen optisch aktiv . Optisch aktive Substanzen kommen paarweise vor optische Antipoden - Isomere, deren physikalische und chemische Eigenschaften unter normalen Bedingungen bis auf eines gleich sind: das Vorzeichen der Drehung der Polarisationsebene. (Wenn einer der optischen Antipoden beispielsweise eine spezifische Drehung von +20 o hat, dann hat der andere eine spezifische Drehung von -20 o).
Projektionsformeln.
Um ein asymmetrisches Atom auf einer Ebene konventionell darzustellen, verwenden Sie Projektionsformeln von E. Fisher . Sie werden erhalten, indem man die Atome, denen das asymmetrische Atom zugeordnet ist, auf eine Ebene projiziert. In diesem Fall wird normalerweise das asymmetrische Atom selbst weggelassen und nur die Schnittlinien und Substituentensymbole bleiben erhalten. Um sich an die räumliche Anordnung der Substituenten zu erinnern, wird in Projektionsformeln häufig eine gestrichelte vertikale Linie beibehalten (die oberen und unteren Substituenten werden über die Zeichenebene hinaus entfernt), dies wird jedoch häufig nicht getan. linkes Modell in der vorherigen Abbildung:

Hier sind einige Beispiele für Projektionsformeln:
(+)-Alanin(-)2-butanol(+)-glycerinaldehyd
Die Namen der Stoffe zeigen ihr Rotationszeichen. Dies bedeutet beispielsweise, dass der linkshändige Antipode von Butanol-2 hat räumliche Konfiguration, genau ausgedrückt durch die obige Formel, und sein Spiegelbild entspricht rechtsdrehendem Butanol-2. Konfigurationsdefinition optische Antipoden wird experimentell durchgeführt.
Im Prinzip lässt sich jeder optische Antipode durch zwölf (!) verschiedene Projektionsformeln abbilden – je nachdem, wo sich das Modell beim Aufbau der Projektion befindet, von welcher Seite wir es betrachten. Um Projektionsformeln zu standardisieren, wurden bestimmte Regeln für deren Schreibweise eingeführt. So wird die Hauptfunktion, wenn sie am Ende der Kette steht, meist oben platziert, die Hauptkette wird vertikal dargestellt.
Um „nicht standardmäßige“ schriftliche Projektionsformeln vergleichen zu können, müssen Sie die folgenden Regeln für die Transformation von Projektionsformeln kennen. Um „nicht standardmäßige“ schriftliche Projektionsformeln vergleichen zu können, müssen Sie die folgenden Regeln für die Transformation von Projektionsformeln kennen.


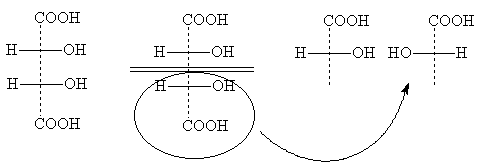
1. Formeln können in der Zeichenebene um 180° gedreht werden, ohne dass sich ihre stereochemische Bedeutung ändert:

2. Zwei (oder eine beliebige gerade Anzahl) Umlagerungen von Substituenten an einem asymmetrischen Atom ändern nichts an der stereochemischen Bedeutung der Formel:

3. Eine (oder eine beliebige ungerade Anzahl) Permutationen von Substituenten am asymmetrischen Zentrum führt zur Formel für den optischen Antipoden:

4. Durch eine 90°-Drehung in der Zeichenebene wird die Formel zu einer antipodalen Formel, sofern nicht gleichzeitig die Bedingung für die Lage der Substituenten relativ zur Zeichenebene geändert wird, d. h. Bedenken Sie nicht, dass sich nun die seitlichen Substituenten hinter der Zeichenebene befinden und die oberen und unteren davor. Wenn Sie eine Formel mit einer gepunkteten Linie verwenden, werden Sie durch die geänderte Ausrichtung der gepunkteten Linie direkt daran erinnert:
5. Anstelle von Permutationen können Projektionsformeln durch Drehen von drei beliebigen Substituenten im oder gegen den Uhrzeigersinn umgewandelt werden; der vierte Substituent ändert seine Position nicht (diese Operation entspricht zwei Permutationen):

6. Projektionsformeln können nicht aus der Zeichenebene abgeleitet werden.
Racemate.
Wenn die Formel einer Substanz ein asymmetrisches Atom enthält, bedeutet dies nicht, dass eine solche Substanz optische Aktivität aufweist. Wenn während einer normalen Reaktion (Substitution in der CH 2 -Gruppe, Addition an einer Doppelbindung usw.) ein asymmetrisches Zentrum entsteht, ist die Wahrscheinlichkeit, dass beide antipodischen Konfigurationen entstehen, gleich. Daher erweist sich die resultierende Substanz trotz der Asymmetrie jedes einzelnen Moleküls als optisch inaktiv. Eine solche optisch inaktive Modifikation, die aus einer gleichen Menge beider Antipoden besteht, nennt man Racemate.
Andere Arten optisch aktiver Substanzen.
In diesem Abschnitt werden einige andere Klassen organischer Verbindungen aufgeführt, die ebenfalls optische Aktivität aufweisen (d. h. als Paare optischer Antipoden vorliegen).

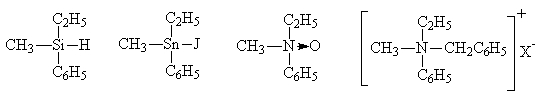
Das Kohlenstoffatom hat kein Monopol auf die Bildung chiraler Zentren in den Molekülen organischer Verbindungen. Das Chiralitätszentrum können auch Silizium-, Zinn- und tetrakovalente Stickstoffatome in quartären Ammoniumsalzen und tertiären Aminoxiden sein:
In diesen Verbindungen hat das Asymmetriezentrum eine tetraedrische Konfiguration, wie das asymmetrische Kohlenstoffatom. Es gibt jedoch auch Verbindungen mit einer anderen räumlichen Struktur des Chiralitätszentrums.

Pyramidenförmig Die Konfiguration besteht aus chiralen Zentren, die aus dreiwertigen Stickstoff-, Phosphor-, Arsen-, Antimon- und Schwefelatomen bestehen. Im Prinzip kann das Asymmetriezentrum als tetraedrisch angesehen werden, wenn das freie Elektronenpaar des Heteroatoms als vierter Substituent angenommen wird:
Es kann auch optische Aktivität auftreten ohne Chiralitätszentrum aufgrund der Chiralität der Struktur des gesamten Moleküls als Ganzes ( molekulare Chiralität oder molekulare Asymmetrie ). Die typischsten Beispiele sind die Anwesenheit chirale Achse oder chirale Ebene .

Die chirale Achse kommt beispielsweise in Allenen vor, die verschiedene Substituenten enthalten S. 2-hybride Kohlenstoffatome. Es ist leicht zu erkennen, dass die folgenden Verbindungen inkompatible Spiegelbilder und daher optische Antipoden sind:

Eine weitere Klasse von Verbindungen mit einer chiralen Achse sind optisch aktive Biphenyle, die über eine chirale Achse verfügen ortho-Positionen haben sperrige Substituenten, die die freie Rotation um die C-C-Bindung, die die Benzolringe verbindet, behindern:
Chirale Ebene zeichnet sich dadurch aus, dass zwischen „oben“ und „unten“ sowie „rechts“ und „links“ unterschieden werden kann. Ein Beispiel für Verbindungen mit einer chiralen Ebene ist die optisch aktive Trance- Cycloocten und optisch aktives Ferrocenderivat:
Diastereomerie.
Verbindungen mit mehreren asymmetrischen Atomen weisen wichtige Merkmale auf, die sie von den zuvor diskutierten einfacheren optisch aktiven Substanzen mit einem Asymmetriezentrum unterscheiden.
Nehmen wir an, dass in einem Molekül einer bestimmten Substanz zwei asymmetrische Atome vorhanden sind; Nennen wir sie bedingt A und B. Es ist leicht zu erkennen, dass Moleküle mit den folgenden Kombinationen möglich sind:
|
((-) |
((-) |
((-) |
((+) |
||
|
Molekül 1 |
F A |
Molekül 3 |
AA |
BB |
|
|
((+) |
((+) |
((+) |
((-) |
||
|
Molekül 2 |
AA |
BB |
Molekül 4 |
AA |
BB |
Die Moleküle 1 und 2 sind ein Paar optischer Antipoden; Gleiches gilt für ein Molekülpaar 3 und 4. Wenn wir Moleküle aus verschiedenen Antipodenpaaren – 1 und 3, 1 und 4, 2 und 3, 2 und 4 – miteinander vergleichen, dann werden wir sehen, dass die aufgeführten Paare sind keine optischen Antipoden: Die Konfiguration eines asymmetrischen Atoms ist gleich, die Konfiguration des anderen ist nicht dieselbe. Das sind Paare Diastereomere , d.h. räumliche Isomere, Nicht bilden optische Antipoden zueinander.
Diastereomere unterscheiden sich nicht nur in der optischen Drehung, sondern auch in allen anderen physikalischen Konstanten: Sie haben unterschiedliche Schmelz- und Siedepunkte, unterschiedliche Löslichkeiten usw. Unterschiede in den Eigenschaften von Diastereomeren sind oft nicht geringer als die Unterschiede in den Eigenschaften zwischen Strukturisomeren .

Ein Beispiel für eine Verbindung dieser Art ist Chloräpfelsäure

Seine stereoisomeren Formen haben die folgenden Projektionsformeln:
erythro-Formen drei- Formen
Titel erythro- Und Trio- kommen von den Namen der Kohlenhydrate Erythrose und Threose. Diese Namen werden verwendet, um die relative Position von Substituenten in Verbindungen mit zwei asymmetrischen Atomen anzugeben: erythro -Isomere Dabei handelt es sich um solche, bei denen in der Standardprojektionsformel auf einer Seite (rechts oder links) zwei identische laterale Substituenten vorkommen. Trio -Isomere haben identische laterale Substituenten auf verschiedenen Seiten der Projektionsformel. Zwei erythro- Isomere sind ein Paar optischer Antipoden; wenn sie gemischt werden, entsteht ein Racemat. Ein Paar optischer Isomere sind und drei- Formen; Sie erzeugen beim Mischen auch ein Racemat, das sich in seinen Eigenschaften vom Racemat unterscheidet erythro- Formen. Somit liegen insgesamt vier optisch aktive Isomere der Chloräpfelsäure und zwei Racemate vor.
Mit einer weiteren Zunahme der Zahl der Asymmetriezentren nimmt die Zahl der räumlichen Isomere zu, und jedes neue Asymmetriezentrum verdoppelt die Zahl der Isomere. Sie wird durch die Formel 2 n bestimmt, wobei n die Anzahl der asymmetrischen Zentren ist.
Die Anzahl der Stereoisomere kann aufgrund teilweiser Symmetrie in einigen Strukturen abnehmen. Ein Beispiel ist Weinsäure, bei der die Zahl der einzelnen Stereoisomere auf drei reduziert ist. Ihre Projektionsformeln:

Formel I ist identisch mit Formel Ia, da sie bei Drehung um 180° in der Zeichenebene in diese übergeht und somit kein neues Stereoisomer darstellt. Diese optisch inaktive Modifikation nennt man Mesoform . Meso- Alle optisch aktiven Substanzen haben Formen mit mehreren identischen (d. h. mit identischen Substituenten verbundenen) Asymmetriezentren. Projektionsformeln Meso- Formen sind immer daran zu erkennen, dass sie durch eine horizontale Linie in zwei Hälften geteilt werden können, die auf Papier zwar formal identisch, in Wirklichkeit aber spiegelverkehrt sind:
Die Formeln II und III stellen die optischen Antipoden der Weinsäure dar; Beim Mischen entsteht ein optisch inaktives Racemat – Traubensäure.
Nomenklatur optischer Isomere.

Das einfachste, älteste, aber immer noch verwendete System der Nomenklatur optischer Antipoden basiert auf dem Vergleich der Projektionsformel des genannten Antipoden mit der Projektionsformel einer bestimmten Standardsubstanz, die als „Schlüssel“ gewählt wurde. Ja fürA-Hydroxysäuren und A-Aminosäuren, der Schlüssel ist der obere Teil ihrer Projektionsformel (in Standardschreibweise):
L-Hydroxysäuren (X = OH) D- Hydroxysäuren (X = OH)
L-Aminosäuren (X = NH 2) D- Aminosäuren (X = NH 2)
Konfiguration allerA-Hydroxysäuren, die in der standardmäßigen schriftlichen Fischer-Projektionsformel links eine Hydroxylgruppe haben, werden mit dem Zeichen gekennzeichnet L; wenn sich das Hydroxyl in der Projektionsformel rechts befindet - Zeichen D

Der Schlüssel zur Bezeichnung der Konfiguration von Zuckern ist Glycerinaldehyd:
L-(-)-Glycerinaldehyd D-(+)-Glycerinaldehyd
Bei Zuckermolekülen die Bezeichnung D- oder L- bezieht sich auf die Konfiguration untere asymmetrisches Zentrum.
System D-,L- Die Bezeichnung weist erhebliche Nachteile auf: Erstens die Bezeichnung D- oder L- gibt die Konfiguration nur eines asymmetrischen Atoms an; zweitens werden für einige Verbindungen unterschiedliche Symbole erhalten, je nachdem, ob der Glycerinaldehyd- oder der Hydroxysäureschlüssel als Schlüssel verwendet wird, zum Beispiel:

Diese Mängel des Schlüsselsystems beschränken seine Verwendung derzeit auf drei Klassen optisch aktiver Substanzen: Zucker, Aminosäuren und Hydroxysäuren. Konzipiert für den allgemeinen Gebrauch R,S-System Kahn, Ingold und Prelog.
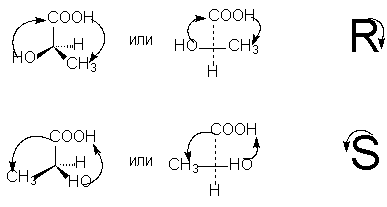
Um die R- bzw. Dann erfolgt die Bewegung während des Übergangs im Kreis der drei verbleibenden Substituenten vom ältesten zum durchschnittlichen Dienstalter und dann zum jüngsten gegen den Uhrzeigersinn - DasS -Isomer (verbunden mit der gleichen Handbewegung beim Schreiben des Buchstabens S), wenn im Uhrzeigersinn - Das R- Isomer (verbunden mit der Bewegung der Hand beim Schreiben des Buchstabens R).
Zur Bestimmung des Dienstalters von Substituenten an einem asymmetrischen Atom werden die Regeln zur Berechnung von Ordnungszahlen verwendet, die wir bereits im Zusammenhang mit der Z,E-Nomenklatur geometrischer Isomere besprochen haben.

Um R-, S-Notationen gemäß der Projektionsformel auszuwählen, ist es notwendig, durch eine gerade Anzahl von Permutationen (die, wie wir wissen, die stereochemische Bedeutung der Formel nicht ändern) die Substituenten so anzuordnen, dass der jüngste von Sie (normalerweise Wasserstoff) stehen am Ende der Projektionsformel. Dann entspricht das Dienstalter der verbleibenden drei Substituenten im Uhrzeigersinn fallend der Bezeichnung R, gegen den Uhrzeigersinn der Bezeichnung S:
5. Methoden zur Gewinnung von Stereoisomeren
Die Gewinnung reiner Stereoisomere ist eine wichtige Aufgabe, da in der Regel nur eine der stereoisomeren Formen biologisch aktiv ist. Unter normalen Bedingungen entstehen mittlerweile in der Regel Gemische von Stereoisomeren – Diastereomere oder optische Antipoden. Um reine stereoisomere Formen zu erhalten, werden diese Gemische verwendet.
II.1. Konformationen (Rotationsisomerie)
Der Übergang vom einfachsten organischen Kohlenwasserstoff, Methan, zu seinem nächsten Homologen, Ethan, wirft Probleme der räumlichen Struktur auf, zu deren Lösung es nicht ausreicht, die im Abschnitt diskutierten Parameter zu kennen. Ohne Änderung der Bindungswinkel oder Bindungslängen kann man sich tatsächlich viele geometrische Formen des Ethanmoleküls vorstellen, die sich voneinander durch die gegenseitige Drehung der Kohlenstofftetraeder um die sie verbindende C-C-Bindung unterscheiden. Als Ergebnis dieser Rotation Rotationsisomere (Konformere) . Die Energie verschiedener Konformere ist nicht gleich, aber die Energiebarriere, die die verschiedenen Rotationsisomere trennt, ist bei den meisten organischen Verbindungen gering. Daher ist es unter normalen Bedingungen in der Regel unmöglich, Moleküle in einer streng definierten Konformation zu fixieren: Normalerweise existieren mehrere Rotationsformen, die sich leicht ineinander umwandeln, im Gleichgewicht nebeneinander.
Die Methoden zur grafischen Darstellung von Konformationen und ihrer Nomenklatur sind wie folgt. Beginnen wir mit dem Ethanmolekül. Dafür kann man die Existenz zweier Konformationen vorhersehen, die sich energetisch maximal unterscheiden. Sie werden unten als dargestellt Perspektivische Projektionen (1) („Sägeziegen“), seitliche Vorsprünge (2 und Newmans Formeln (3).
In der perspektivischen Projektion (1a, 1b) muss man sich die C-C-Verbindung in die Ferne gehend vorstellen; Das Kohlenstoffatom auf der linken Seite befindet sich in der Nähe des Beobachters und das Kohlenstoffatom auf der rechten Seite ist weiter von ihm entfernt.
In der seitlichen Projektion (2a, 2b) liegen vier H-Atome in der Zeichenebene; Tatsächlich erstrecken sich die Kohlenstoffatome etwas aus dieser Ebene heraus, aber vereinfacht wird üblicherweise davon ausgegangen, dass sie auch in der Zeichenebene liegen. „Fett“ keilförmige Bindungen durch die Verdickung des Keils deuten auf einen Austritt aus der Ebene in Richtung des Beobachters des Atoms hin, dem die Verdickung zugewandt ist. Gestrichelte keilförmige Verbindungen markieren den Abstand zum Betrachter.
In der Newman-Projektion (3a, 3b) wird das Molekül entlang der C-C-Bindung betrachtet (in der durch den Pfeil in den Formeln 1a, b angegebenen Richtung). Drei Linien, die in einem Winkel von 120° vom Mittelpunkt des Kreises auseinanderlaufen, zeigen die Bindungen des Kohlenstoffatoms an, das dem Betrachter am nächsten ist; Die Linien, die hinter dem Kreis „herausragen“, sind die Bindungen des entfernten Kohlenstoffatoms.
Die links dargestellte Konformation wird aufgerufen verdeckt : Dieser Name erinnert uns daran, dass die Wasserstoffatome beider CH 3 -Gruppen einander gegenüberstehen. Die verfinsterte Konformation hat eine erhöhte innere Energie und ist daher ungünstig. Die rechts dargestellte Konformation wird aufgerufen gehemmt , was bedeutet, dass die freie Rotation um die C-C-Bindung in dieser Position „gehemmt“ ist, d. h. das Molekül liegt überwiegend in dieser Konformation vor.
Die minimale Energie, die erforderlich ist, um ein Molekül vollständig um eine bestimmte Bindung zu drehen, wird aufgerufen Rotationsbarriere für diese Verbindung. Die Rotationsbarriere in einem Molekül wie Ethan kann als Änderung der potentiellen Energie des Moleküls als Funktion der Änderung ausgedrückt werden Diederwinkel (Torsionswinkel). Systeme. Der Diederwinkel (bezeichnet als Tau) ist in der folgenden Abbildung dargestellt:
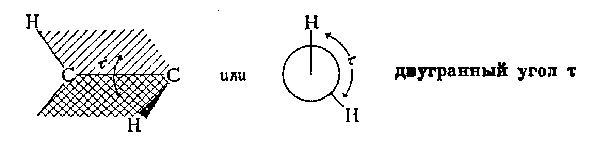
Das Energieprofil der Rotation um die C-C-Bindung in Ethan ist in der folgenden Abbildung dargestellt. Die Drehung des „hinteren“ Kohlenstoffatoms wird durch die Änderung des Diederwinkels zwischen den beiden dargestellten Wasserstoffatomen dargestellt. Der Einfachheit halber wurden die restlichen Wasserstoffatome weggelassen. Die Rotationsbarriere, die die beiden Formen von Ethan trennt, beträgt nur 3 kcal/mol (12,6 kJ/mol). Die Minima der Potentialenergiekurve entsprechen inhibierten Konformationen und die Maxima entsprechen okkludierten Konformationen. Da bei Raumtemperatur die Energie einiger molekularer Kollisionen 20 kcal/mol (ca. 80 kJ/mol) erreichen kann, kann diese Barriere von 12,6 kJ/mol leicht überwunden werden und die Rotation in Ethan wird als frei angesehen.

Wir betonen, dass jeder Punkt auf der potentiellen Energiekurve einer bestimmten Konformation entspricht. Die den Minima entsprechenden Punkte entsprechen Konformationsisomeren die vorherrschenden Komponenten in einer Mischung aller möglichen Konformationen .
Je komplexer das Molekül wird, desto mehr mögliche Konformationen nehmen zu, die sich energetisch deutlich unterscheiden. Ja für N-Butan kann bereits in sechs Konformationen dargestellt werden, die sich in der relativen Anordnung der CH 3 -Gruppen unterscheiden, d. h. Drehung um die zentrale Verbindung C-C. Nachfolgend sind die Konformationen von n-Butan als Newman-Projektionen dargestellt. Die links dargestellten Konformationen (schattiert) sind energetisch ungünstig; nur gehemmte werden praktisch realisiert.

Die verschiedenen verfinsterten und gehemmten Konformationen von Butan sind energetisch nicht gleich. Nachfolgend sind die entsprechenden Energien aller Konformationen dargestellt, die sich bei der Rotation um die zentrale C-C-Bindung bilden:



Je komplexer ein Molekül wird, desto mehr mögliche Konformationen gibt es.
Konformationen sind also verschiedene nichtidentische räumliche Formen eines Moleküls, die eine bestimmte Konfiguration aufweisen. Konformere sind stereoisomere Strukturen, die sich im mobilen Gleichgewicht befinden und durch Rotation um einfache Bindungen ineinander umgewandelt werden können.
Manchmal wird die Barriere für solche Umwandlungen hoch genug, um stereoisomere Formen zu trennen (Beispiel: optisch aktive Biphenyle). In solchen Fällen spricht man nicht mehr von Konformeren, sondern von tatsächlich existierenden Stereoisomere .
II.2. Geometrische Isomerie
Eine wichtige Konsequenz der Starrheit einer Doppelbindung (das Fehlen einer Rotation um sie herum) ist ihre Existenz geometrische Isomere . Die häufigsten davon sind cis-trans-Isomere Verbindungen der Ethylenreihe mit ungleichen Substituenten an ungesättigten Atomen. Das einfachste Beispiel sind die Isomere von Buten-2.

Geometrische Isomere haben die gleiche chemische Struktur (die gleiche Reihenfolge der chemischen Bindungen) und unterscheiden sich in der räumlichen Anordnung der Atome Konfigurationen . Dieser Unterschied führt zu einem Unterschied in den physikalischen (sowie chemischen) Eigenschaften. Geometrische Isomere können im Gegensatz zu Konformeren in reiner Form isoliert werden und liegen als einzelne, stabile Substanzen vor. Ihre gegenseitige Umwandlung erfordert normalerweise Energie in der Größenordnung von 125–170 kJ/mol (30–40 kcal/mol). Diese Energie kann durch Erhitzen oder Bestrahlung zugeführt werden.
Im einfachsten Fall bereitet die Nomenklatur geometrischer Isomere keine Schwierigkeiten: cis- Formen sind geometrische Isomere, bei denen identische Substituenten auf derselben Seite der Pi-Bindungsebene liegen. Trance- Isomere haben identische Substituenten auf verschiedenen Seiten der Pi-Bindungsebene. In komplexeren Fällen wird es verwendet Z,E-Nomenklatur . Sein Hauptprinzip: die Konfiguration angeben cis-(Z, aus dem Deutschen Zusammen – zusammen) oder Trance-(E, vom Deutschen Entgegen – gegenüber) Standort hochrangige Stellvertreter mit einer Doppelbindung.
Im Z,E-System gelten Substituenten mit einer höheren Ordnungszahl als höher. Wenn die Atome, die direkt mit ungesättigten Kohlenstoffen verbunden sind, gleich sind, wandern sie bei Bedarf in die „zweite Schicht“ – in die „dritte Schicht“ usw.
Betrachten wir die Anwendung der Regeln der Z,E-Nomenklatur anhand von zwei Beispielen.

| ICH | II |
Beginnen wir mit Formel I, bei der alles durch die Atome der „ersten Schicht“ gelöst wird. Nachdem wir ihre Ordnungszahlen geordnet haben, stellen wir fest, dass die älteren Substituenten jedes Paares (Brom im oberen Teil der Formel und Stickstoff im unteren Teil) in sind Trance-Position, daher die stereochemische Bezeichnung E:
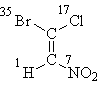 E-1-Brom-1-chlor-2-nitroethen
E-1-Brom-1-chlor-2-nitroethen
Um die stereochemische Bezeichnung der Struktur II zu bestimmen, ist es notwendig, nach Unterschieden in den „höheren Schichten“ zu suchen. In der ersten Schicht unterscheiden sich die Gruppen CH 3, C 2 H 5, C 3 H 7 nicht. In der zweiten Schicht hat die CH 3 -Gruppe eine Summe der Ordnungszahlen von drei (drei Wasserstoffatome), während die C 2 H 5- und C 3 H 7-Gruppen jeweils 8 haben. Dies bedeutet, dass die CH 3-Gruppe nicht berücksichtigt wird - es ist jünger als die anderen beiden. Somit sind die älteren Gruppen C 2 H 5 und C 3 H 7, es ist in cis-Position; stereochemische Bezeichnung Z.
 Z-3-Methylhepten-3
Z-3-Methylhepten-3
Müsste festgestellt werden, welche Gruppe älter ist – C 2 H 5 oder C 3 H 7 –, müsste man zu den Atomen der „dritten Schicht“ gehen; die Summe der Ordnungszahlen in dieser Schicht wäre für beide Gruppen gleich auf 3 bzw. 8, d.h. C3H7 ist älter als C2H5. In komplexeren Fällen der Vorrangbestimmung müssen zusätzliche Bedingungen berücksichtigt werden, wie zum Beispiel: Ein durch eine Doppelbindung verbundenes Atom wird zweimal gezählt, ein durch eine Dreifachbindung verbundenes Atom – dreimal; Von den Isotopen ist das schwerere älter (Deuterium ist älter als Wasserstoff) und einige andere.
Beachten Sie, dass die Notation Z Nicht sind Synonyme cis- Bezeichnungen, wie auch Bezeichnungen E, entsprechen nicht immer dem Ort Trance-, Zum Beispiel:

cis- 1,2-Dichlorpropen-1 cis- 1,2-Dichlor-1-brompropen-1
Z-1,2-Dichlorpropen-1 E-1,2-Dichlor-1-brompropen-1
Kontrollaufgaben
1. Bombicol – ein Pheromon (sexueller Lockstoff) der Seidenraupe – ist E-10-Z-12-Hexadecadienol-1. Zeichnen Sie seine Strukturformel.
2. Benennen Sie die folgenden Verbindungen mithilfe der Z,E-Nomenklatur:

II.3. Optische Isomerie (Enantiomerie)
Unter den organischen Verbindungen gibt es Stoffe, die die Polarisationsebene des Lichts drehen können. Dieses Phänomen wird als optische Aktivität bezeichnet, und die entsprechenden Substanzen sind es auch optisch aktiv . Optisch aktive Substanzen kommen paarweise vor optische Antipoden - Isomere, deren physikalische und chemische Eigenschaften unter normalen Bedingungen identisch sind, mit Ausnahme einer Sache – dem Vorzeichen der Drehung der Polarisationsebene. (Wenn einer der optischen Antipoden beispielsweise eine spezifische Drehung [ANMERKUNG 1] +20 o hat, dann hat der andere eine spezifische Drehung von -20 o).
II.4. Projektionsformeln
Um ein asymmetrisches Atom auf einer Ebene konventionell darzustellen, verwenden Sie Projektionsformeln von E. Fisher . Sie werden erhalten, indem man die Atome, denen das asymmetrische Atom zugeordnet ist, auf eine Ebene projiziert. In diesem Fall wird normalerweise das asymmetrische Atom selbst weggelassen und nur die Schnittlinien und Substituentensymbole bleiben erhalten. Um sich an die räumliche Anordnung der Substituenten zu erinnern, wird in Projektionsformeln häufig eine gestrichelte vertikale Linie beibehalten (die oberen und unteren Substituenten werden über die Zeichenebene hinaus entfernt), dies wird jedoch häufig nicht getan. Im Folgenden finden Sie verschiedene Möglichkeiten, die Projektionsformel entsprechend dem linken Modell in der vorherigen Abbildung zu schreiben:
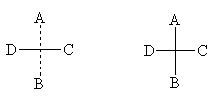
Hier sind einige Beispiele für Projektionsformeln:

(+)-Alanin (-)-Butanol (+)-Glycerinaldehyd
Die Namen von Stoffen geben ihr Rotationszeichen an: Dies bedeutet beispielsweise, dass der linkshändige Antipode von Butanol-2 hat räumliche Konfiguration , genau ausgedrückt durch die obige Formel, und sein Spiegelbild entspricht rechtsdrehendem Butanol-2. Konfigurationsdefinition optische Antipoden wird experimentell durchgeführt [ANMERKUNG 3].
Im Prinzip lässt sich jeder optische Antipode durch zwölf (!) verschiedene Projektionsformeln abbilden – je nachdem, wie das Modell bei der Projektion positioniert ist und von welcher Seite wir es betrachten. Um Projektionsformeln zu standardisieren, wurden bestimmte Regeln für deren Schreibweise eingeführt. So wird die Hauptfunktion, wenn sie am Ende der Kette steht, meist oben platziert, die Hauptkette wird vertikal dargestellt.
Um „nicht standardmäßige“ schriftliche Projektionsformeln vergleichen zu können, müssen Sie die folgenden Regeln für die Transformation von Projektionsformeln kennen.
1. Formeln können in der Zeichenebene um 180° gedreht werden, ohne dass sich ihre stereochemische Bedeutung ändert:

2. Zwei (oder eine beliebige gerade Anzahl) Umlagerungen von Substituenten an einem asymmetrischen Atom ändern nichts an der stereochemischen Bedeutung der Formel:

3. Eine (oder eine beliebige ungerade Anzahl) Permutationen von Substituenten am asymmetrischen Zentrum führt zur Formel für den optischen Antipoden:
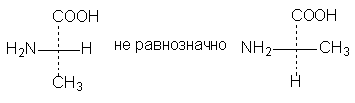
4. Durch eine 90°-Drehung in der Zeichenebene wird die Formel zu einer antipodalen Formel, sofern nicht gleichzeitig die Bedingung für die Lage der Substituenten relativ zur Zeichenebene geändert wird, d. h. Bedenken Sie nicht, dass sich nun die seitlichen Substituenten hinter der Zeichenebene befinden und die oberen und unteren davor. Wenn Sie eine Formel mit einer gepunkteten Linie verwenden, werden Sie durch die geänderte Ausrichtung der gepunkteten Linie direkt daran erinnert:

5. Anstelle von Permutationen können Projektionsformeln durch Drehen von drei beliebigen Substituenten im oder gegen den Uhrzeigersinn umgewandelt werden; der vierte Substituent ändert seine Position nicht (diese Operation entspricht zwei Permutationen):

6. Projektionsformeln können nicht aus der Zeichenebene abgeleitet werden (d. h. man kann sie beispielsweise nicht „im Licht“ von der Rückseite des Papiers aus betrachten – in diesem Fall ändert sich die stereochemische Bedeutung der Formel).
II.5. Racemate
Wenn die Formel einer Substanz ein asymmetrisches Atom enthält, bedeutet dies nicht, dass eine solche Substanz optische Aktivität aufweist. Wenn während einer normalen Reaktion (Substitution in der CH 2 -Gruppe, Addition an einer Doppelbindung usw.) ein asymmetrisches Zentrum entsteht, ist die Wahrscheinlichkeit, dass beide antipodischen Konfigurationen entstehen, gleich. Daher erweist sich die resultierende Substanz trotz der Asymmetrie jedes einzelnen Moleküls als optisch inaktiv. Eine solche optisch inaktive Modifikation, die aus einer gleichen Menge beider Antipoden besteht, nennt man Racemate [ANMERKUNG 4] .
II.6. Diastereomerie
Verbindungen mit mehreren asymmetrischen Atomen weisen wichtige Merkmale auf, die sie von den zuvor diskutierten einfacheren optisch aktiven Substanzen mit einem Asymmetriezentrum unterscheiden.
Nehmen wir an, dass in einem Molekül einer bestimmten Substanz zwei asymmetrische Atome vorhanden sind; Nennen wir sie bedingt A und B. Es ist leicht zu erkennen, dass Moleküle mit den folgenden Kombinationen möglich sind:
Die Moleküle 1 und 2 sind ein Paar optischer Antipoden; Gleiches gilt für ein Molekülpaar 3 und 4. Wenn wir Moleküle aus verschiedenen Antipodenpaaren – 1 und 3, 1 und 4, 2 und 3, 2 und 4 – miteinander vergleichen, dann werden wir sehen, dass die aufgeführten Paare sind keine optischen Antipoden: Die Konfiguration eines asymmetrischen Atoms ist gleich, die Konfiguration des anderen ist nicht dieselbe. Das sind alles Paare Diastereomere , d.h. räumliche Isomere, Nicht bilden optische Antipoden zueinander.
Diastereomere unterscheiden sich nicht nur in der optischen Drehung, sondern auch in allen anderen physikalischen Konstanten: Sie haben unterschiedliche Schmelz- und Siedepunkte, unterschiedliche Löslichkeiten usw. Unterschiede in den Eigenschaften von Diastereomeren sind oft nicht geringer als die Unterschiede in den Eigenschaften zwischen Strukturisomeren .
Ein Beispiel für eine Verbindung dieser Art ist Chloräpfelsäure

Seine stereoisomeren Formen haben die folgenden Projektionsformeln:

erythro- Formen drei- Formen
Titel erythro- Und Trio- kommen von den Namen der Kohlenhydrate Erythrose und Threose. Diese Namen werden verwendet, um die relative Position von Substituenten in Verbindungen mit zwei asymmetrischen Atomen anzugeben: erythro -Isomere Dabei handelt es sich um solche, bei denen in der Standardprojektionsformel auf einer Seite (rechts oder links) zwei identische laterale Substituenten vorkommen. Trio -Isomere haben identische laterale Substituenten auf verschiedenen Seiten der Projektionsformel [ANMERKUNG 5].
Zwei erythro- Isomere sind ein Paar optischer Antipoden; wenn sie gemischt werden, entsteht ein Racemat. Ein Paar optischer Isomere sind und drei- Formen; Sie erzeugen beim Mischen auch ein Racemat, das sich in seinen Eigenschaften vom Racemat unterscheidet erythro- Formen. Somit liegen insgesamt vier optisch aktive Isomere der Chloräpfelsäure und zwei Racemate vor.
Mit einer weiteren Zunahme der Zahl der Asymmetriezentren nimmt die Zahl der räumlichen Isomere zu, und jedes neue Asymmetriezentrum verdoppelt die Zahl der Isomere. Sie wird durch die Formel 2 n bestimmt, wobei n die Anzahl der asymmetrischen Zentren ist.
Die Anzahl der Stereoisomere kann aufgrund teilweiser Symmetrie in einigen Strukturen abnehmen. Ein Beispiel ist Weinsäure, bei der die Zahl der einzelnen Stereoisomere auf drei reduziert ist. Ihre Projektionsformeln:

Formel I ist identisch mit Formel Ia: Sie wandelt sich bei einer Drehung um 180° in der Zeichenebene in diese um und stellt daher kein neues Stereoisomer dar. Dies ist eine optisch inaktive Modifikation - Mesoform . Im Gegensatz zu einem Racemat, das in optische gespalten werden kann Antipoden, Meso- Die Form ist grundsätzlich unzerstörbar: Jedes seiner Moleküle hat ein asymmetrisches Zentrum einer Konfiguration und das zweite das Gegenteil. Dadurch kommt es zu einer intramolekularen Kompensation der Rotation beider Asymmetriezentren.
Meso- Alle optisch aktiven Substanzen haben Formen mit mehreren identischen (d. h. mit identischen Substituenten assoziierten) Asymmetriezentren [ANMERKUNG 6]. Projektionsformeln Meso- Formen sind immer daran zu erkennen, dass sie durch eine horizontale Linie immer in zwei Hälften geteilt werden können, die auf Papier zwar formal identisch, in Wirklichkeit aber spiegelverkehrt sind:
Die Formeln II und III stellen die optischen Antipoden der Weinsäure dar; Beim Mischen entsteht ein optisch inaktives Racemat – Traubensäure.
II.7. Nomenklatur optischer Isomere
Das einfachste, älteste, aber immer noch verwendete System der Nomenklatur optischer Antipoden basiert auf dem Vergleich der Projektionsformel des genannten Antipoden mit der Projektionsformel einer bestimmten Standardsubstanz, die als „Schlüssel“ gewählt wurde. Für Alpha-Hydroxysäuren und Alpha-Aminosäuren ist der Schlüssel daher der obere Teil ihrer Projektionsformel (in Standardschreibweise):

L- Hydroxysäuren (X = OH) D- Hydroxysäuren (X = OH)
L-Aminosäuren (X = NH 2) D- Aminosäuren (X = NH 2)
Die Konfiguration aller Alpha-Hydroxysäuren, die in der Standardformel der geschriebenen Fischer-Projektion links eine Hydroxylgruppe haben, wird durch das Vorzeichen angezeigt L; wenn sich das Hydroxyl in der Projektionsformel rechts befindet - Zeichen D[ANMERKUNG 7] .
Der Schlüssel zur Bezeichnung der Konfiguration von Zuckern ist Glycerinaldehyd:
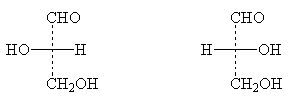
L-(-)-Glycerinaldehyd D-(+)-Glycerinaldehyd
Bei Zuckermolekülen die Bezeichnung D- oder L- bezieht sich auf die Konfiguration untere asymmetrisches Zentrum.
System D-,L- Die Bezeichnung weist erhebliche Nachteile auf: Erstens die Bezeichnung D- oder L- gibt die Konfiguration nur eines asymmetrischen Atoms an; zweitens werden für einige Verbindungen unterschiedliche Symbole erhalten, je nachdem, ob der Glycerinaldehyd- oder der Hydroxysäureschlüssel als Schlüssel verwendet wird, zum Beispiel:

Diese Mängel des Schlüsselsystems beschränken seine Verwendung derzeit auf drei Klassen optisch aktiver Substanzen: Zucker, Aminosäuren und Hydroxysäuren. Konzipiert für den allgemeinen Gebrauch „R,S-System Kahn, Ingold und Prelog [ANMERKUNG 8].
Um die R- bzw. Dann erfolgt die Bewegung während des Übergangs im Kreis der drei verbleibenden Substituenten vom ältesten zum durchschnittlichen Dienstalter und dann zum jüngsten gegen den Uhrzeigersinn - Das R -Isomer (verbunden mit der gleichen Handbewegung beim Schreiben des Buchstabens R), wenn im Uhrzeigersinn - Das S- Isomer (verbunden mit der gleichen Handbewegung beim Schreiben des Buchstabens S).
Um das Dienstalter von Substituenten an einem asymmetrischen Atom zu bestimmen, werden die Regeln zur Berechnung von Ordnungszahlen verwendet, die wir bereits im Zusammenhang mit der Z,E-Nomenklatur geometrischer Isomere betrachtet haben (siehe).
Um R-, S-Notationen gemäß der Projektionsformel auszuwählen, ist es notwendig, durch eine gerade Anzahl von Permutationen (die, wie wir wissen, die stereochemische Bedeutung der Formel nicht ändern) die Substituenten so anzuordnen, dass der jüngste von Sie (normalerweise Wasserstoff) stehen am Ende der Projektionsformel. Dann entspricht das Dienstalter der verbleibenden drei Substituenten im Uhrzeigersinn fallend der Bezeichnung R, gegen den Uhrzeigersinn der Bezeichnung S [ANMERKUNG 9]:
Kontrollaufgaben
3. Bestimmen Sie die Konfiguration des asymmetrischen Zentrums von Ascorbinsäure (Vitamin C) (durch R,S-Nomenklatur und im Vergleich zu Glycerinaldehyd):

4. Das Alkaloid Ephedrin hat die Formel:
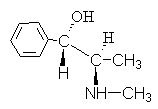
Geben Sie einen Namen für diese Verbindung ein R,S- Nomenklatur.
5. Cystein – eine nicht essentielle Aminosäure, die an der Regulierung von Stoffwechselprozessen beteiligt ist L-1-Amino-2-mercaptopropionsäure. Zeichnen Sie seine Strukturformel und geben Sie seinen Namen an R,S- Nomenklatur.
6. Levomycetin (Breitbandantibiotikum) ist D(-)-threo-1-para-nitrophenyl-2-dichloracetylaminopropandiol-1,3. Zeichnen Sie seine Struktur in Form der Fischer-Projektionsformel.
7. Synestrol ist ein synthetisches Östrogenmedikament mit nichtsteroidaler Struktur. Geben Sie den Namen an, der auf die stereochemische Konfiguration hinweist:
II.8. Stereochemie zyklischer Verbindungen
Wenn eine Kette von Kohlenstoffatomen zu einem planaren Zyklus geschlossen wird, müssen die Bindungswinkel der Kohlenstoffatome von ihrem normalen Tetraederwert abweichen, und das Ausmaß dieser Abweichung hängt von der Anzahl der Atome im Zyklus ab. Je größer der Abweichungswinkel der Valenzbindungen ist, desto größer ist die Energiereserve des Moleküls, desto instabiler ist der Zyklus. Allerdings hat nur ein dreigliedriger zyklischer Kohlenwasserstoff (Cyclopropan) eine flache Struktur; Ausgehend von Cyclobutan haben Cycloalkanmoleküle eine nichtplanare Struktur, die die „Spannung“ im System verringert.
Das Cyclohexanmolekül kann in mehreren Konformationen vorliegen, in denen die „normalen“ Bindungswinkel erhalten bleiben (der Einfachheit halber sind nur Kohlenstoffatome dargestellt):
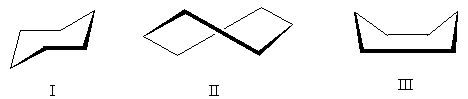
Die energetisch günstigste Konformation ist die Konformation I – die sogenannte Form "Sessel". Konformation II - "Twist" - nimmt eine Zwischenstellung ein: Sie ist weniger günstig als die Sesselkonformation (aufgrund des Vorhandenseins verdeckter Wasserstoffatome), aber günstiger als die Konformation III. Konformation III - "Bad" - das ungünstigste der drei aufgrund der erheblichen Abstoßung nach oben gerichteter Wasserstoffatome.
Die Betrachtung der zwölf C-H-Bindungen in der Sesselkonformation ermöglicht es uns, sie in zwei Gruppen zu unterteilen: sechs axial abwechselnd nach oben und unten gerichtete Verbindungen und sechs Äquatorial Anschlüsse nach den Seiten gerichtet. In einfach substituierten Cyclohexanen kann sich der Substituent entweder in äquatorialer oder axialer Position befinden. Diese beiden Konformationen befinden sich normalerweise im Gleichgewicht und gehen durch die Twist-Konformation schnell ineinander über:
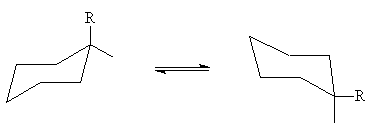
Die äquatoriale Konformation (e) ist meist energieärmer und daher vorteilhafter als die axiale Konformation (a).
Wenn Substituenten (Seitenketten) in Ringen auftreten, steht der Forscher neben dem Problem der Konformation des Rings selbst auch vor Problemen Substituentenkonfigurationen : also bei Vorliegen zweier gleicher oder verschiedener Substituenten, cis-trans-Isomer. Beachten Sie, dass darüber gesprochen wird cis-trans-Konfiguration von Substituenten ist nur sinnvoll, wenn sie auf gesättigte kleine und mittlere Ringe (bis C 8) angewendet wird: In Ringen mit einer großen Anzahl von Einheiten wird die Mobilität so wichtig, dass Diskussionen darüber geführt werden cis- oder Trance- Die Positionen der Abgeordneten verlieren ihre Bedeutung.
Ein klassisches Beispiel sind stereoisomere Cyclopropan-1,2-dicarbonsäuren. Es gibt zwei stereoisomere Säuren: eine davon mit einem Schmp. 139 o C, ist in der Lage, zyklisches Anhydrid zu bilden und ist daher cis-Isomer. Eine weitere stereoisomere Säure, Schmp. 175 °C, bildet kein zyklisches Anhydrid; Das Trance-Isomer [ANMERKUNG 10]:

Zwei stereoisomere 1,2,2-Trimethylcyclopentan-1,3-dicarbonsäuren stehen im gleichen Verhältnis zueinander. Eine davon, Kampfersäure, Schmp. 187 o C, bildet ein Anhydrid und ist daher cis-Isomer. Die andere ist Isocamphersäure, Schmp. 171 o C, - bildet dabei kein Anhydrid Trance-Isomer:

cis-trans-
Obwohl das Cyclopentanmolekül nicht wirklich planar ist, ist es der Übersichtlichkeit halber zweckmäßig, es in flacher Form darzustellen, wie in der Abbildung oben, wobei zu berücksichtigen ist, dass in cis- Isomer hat zwei Substituenten auf einer Seite des Zyklus , und in Trance-Isomer - auf gegenüberliegenden Seiten des Zyklus .
Disubstituierte Cyclohexan-Derivate können auch in cis- oder trans-Form vorliegen:
Das Kohlenstoffatom hat kein Monopol auf die Bildung chiraler Zentren in den Molekülen organischer Verbindungen. Das Chiralitätszentrum können auch Silizium-, Zinn- und tetrakovalente Stickstoffatome in quartären Ammoniumsalzen und tertiären Aminoxiden sein:
In diesen Verbindungen hat das Asymmetriezentrum eine tetraedrische Konfiguration, wie das asymmetrische Kohlenstoffatom. Es gibt jedoch auch Verbindungen mit einer anderen räumlichen Struktur des Chiralitätszentrums.
Chiralitätszentren, die aus Atomen des dreiwertigen Stickstoffs, Phosphors, Arsens, Antimons und Schwefels bestehen, haben eine Pyramidenkonfiguration. Im Prinzip kann das Asymmetriezentrum als tetraedrisch angesehen werden, wenn das freie Elektronenpaar des Heteroatoms als vierter Substituent angenommen wird:

Es kann auch optische Aktivität auftreten ohne Chiralitätszentrum aufgrund der Chiralität der Struktur des gesamten Moleküls als Ganzes ( molekulare Chiralität oder molekulare Asymmetrie ). Die typischsten Beispiele sind die Anwesenheit chirale Achse oder chirale Ebene .
Die chirale Achse kommt beispielsweise in Allenen vor, die verschiedene Substituenten enthalten S. 2-hybride Kohlenstoffatome. Es ist leicht zu erkennen, dass die folgenden Verbindungen Spiegelbilder und daher optische Antipoden sind:

Die Chiralitätsachse ist in den Abbildungen durch einen Pfeil dargestellt.
Eine weitere Klasse von Verbindungen mit einer chiralen Achse sind optisch aktive Biphenyle, die über eine chirale Achse verfügen ortho-Positionen haben sperrige Substituenten, die die freie Rotation um die C-C-Bindung, die die Arenringe verbindet, behindern:

Chirale Ebene zeichnet sich dadurch aus, dass zwischen „oben“ und „unten“ sowie „rechts“ und „links“ unterschieden werden kann. Ein Beispiel für Verbindungen mit einer chiralen Ebene ist die optisch aktive Trance- Cycloocten und ein optisch aktives Ferrocenderivat.





