Die Hämoglobinsynthese erfolgt durch die synchrone Produktion von Häm- und Globin-Polypeptidketten mit anschließender Bildung eines vollständigen Moleküls. Aminosäuren sind das Substrat für die Bildung von Globin. An der Synthese von gem. Die Hämoglobinsynthese beginnt in Normozyten. Mit der weiteren Reifung der Erythrozytenzelle, einer Abnahme der Anzahl der Polysomen im Zytoplasma, nimmt auch die Hämoglobinsynthese ab. In Retikulozyten ist die Hämoglobinsynthese noch auf ribosomal-zytoplasmatischer Ebene möglich. Reife rote Blutkörperchen synthetisieren kein Hämoglobin.
Der Prozess der Hämoglobinsynthese während der Erythropoese ist mit dem Verbrauch von körpereigenem Eisen verbunden. Folgende Proteinverbindungen spielen eine wichtige Rolle beim Austausch von körpereigenem Eisen: Transferrin (Siderophilin), Ferritin und Hämosiderin.
Transferrin- ein im Blutplasma enthaltenes spezifisches Protein ist β-Globulin mit Molekulargewicht etwa 80.000 D. Es übernimmt eine Transportfunktion und sorgt für den Transport von Eisen aus der Darmschleimhaut und den Nebenhöhlen des Milzparenchyms in das Knochenmark, wo es für die Erythropoese verwertet wird.
Ferritin- ein wasserlöslicher Komplex von Eisenhydroxid mit dem Protein Apoferritin. Das Molekulargewicht von Ferritin beträgt ca. 460.000 D, der Eisengehalt beträgt ca. 20 % seines Gewichts.
Hämosiderin eine ähnliche Zusammensetzung wie Ferritin hat, beträgt der Eisengehalt darin etwa 30% der Gesamtmasse des Hämosiderinmoleküls. Die Hauptablagerungsorte von Hämosiderin sind das Knochenmark, die Leber und die Milz.
Der Körper eines gesunden Erwachsenen enthält im Allgemeinen etwa 3-5 g körpereigenes Eisen, und der Erythronfonds enthält etwa 60-70% und die Eisenreserven (Ferritin und Hämosiderin der inneren Organe) betragen 30-40%. Transferrin enthält etwa 3-4 mg Eisen, Enzyme verschiedener Organe und Gewebe enthalten etwa 150 mg Eisen.
Der Gehalt an körpereigenem Eisen wird maßgeblich durch die Konstanz der Aufnahme von exogenem Eisen bestimmt. Dieser Prozess ist jedoch streng limitiert; die im Laufe des Tages aus der Nahrung aufgenommene Eisenmenge beträgt auch bei stark erhöhtem Bedarf 2,0-2,5 mg nicht. Von großer Bedeutung ist nicht nur die Menge an Eisen in einem bestimmten Produkt, sondern auch die Form seines Gehalts und dementsprechend die Möglichkeit seiner Aufnahme aus diesem Produkt. Eisen kommt in vielen Lebensmitteln vor, sowohl in pflanzlichen als auch in tierischen Quellen. Fleisch, Leber, Nieren, Hülsenfrüchte, getrocknete Aprikosen, Pflaumen, Rosinen, Reis, Brot, Äpfel enthalten viel Eisen. Aus Reis wird jedoch nicht mehr als 1 % Eisen und aus Früchten nicht mehr als 3 % aufgenommen. Aus Rindfleisch und insbesondere Kalbfleisch wird viel Eisen aufgenommen - bis zu 22%, aus Fisch - bis zu 11%.
Lebensmittel können verschiedene Formen von Eisen enthalten, das Teil von Häm, Ferritin, Hämosiderin, Komplexverbindungen mit Oxalaten und Phosphaten ist.
Eisen, das Bestandteil hämhaltiger Verbindungen ist, wird absorbiert
viel besser als Ferritin und Hämosiderin.
Dem Magenfaktor, insbesondere der normalen HCl-Sekretion, wird bei der Regulation der Aufnahme von in der Nahrung enthaltenem Eisen in Form einer dreiwertigen Verbindung nur eine Hilfsfunktion zugeschrieben. Die Aufnahme von Eisen in zweiwertiger Form, einschließlich desjenigen, das Teil des Häms ist, hängt praktisch nicht vom Zustand der Sekretionskapazität des Magens ab. Es hat sich gezeigt, dass auch bei Achilien die Eisenaufnahme recht zufriedenstellend ist. Dieser Standpunkt kann jedoch nicht als allgemein akzeptiert gelten, da Salzsäure nach anderen Angaben Eiseneisen im Magen-Darm-Trakt stabilisiert, die Bildung leicht assimilierbarer Eisenkomplexverbindungen fördert.
Die Aktivierung der Prozesse der Eisenaufnahme aus dem Darm erfolgt bei Hypoxie, erhöhter Erythropoese und Abnahme der Eisenkonzentration im Blutplasma. Die Eisenaufnahme wird durch den Einfluss von Ascorbin-, Bernstein-, Brenztraubensäure, Fructose, Sorbit und Alkohol verstärkt.
In der Darmschleimhaut befindet sich ein Enzym Häm-Oxygenase wird für den Abbau des Hämmoleküls in Bilirubin, Kohlenmonoxid und ionisiertes Eisen benötigt. Auf der Oberfläche von Enterozyten befindet sich ein spezifisches Rezeptorprotein Anoferritin, das für die Eisenbindung, seinen Eintritt in Enterozyten und die Bildung einer labilen Form der Eisenablagerung im Epithel der Darmschleimhaut sorgt. Es ist zu beachten, dass nur Eisen(II) im Darm resorbiert wird, und wenn die Konzentration von Eisen(II) im Darm stark ansteigt, nimmt der Resorptionsprozess entsprechend zu. Eisen (III) wird im Darm praktisch nicht resorbiert.
Der Hauptort der Eisenablagerung ist die Leber, und die Ablagerungsformen sind Ferritin und Hämosiderin.
Der Eisengehalt im Blutserum weist unter normalen Bedingungen eine große Schwankungsbreite auf - von 70 bis 170 µg% (12,5-30,4 µmol / l). Die Eisenbindungskapazität von Blutserum reicht normalerweise von 30,6 bis 84,6 µmol/l (70-470 µg/%). Unter der Eisenbindungskapazität des Blutserums versteht man die Menge an Eisen, die an Transferrin binden kann.
Der Eisenverlust aus dem Körper erfolgt auf verschiedene Weise: mit Kot, Urin, Schweiß, Hautepithel, und etwa 0,1 mg Eisen gehen mit dem Urin verloren, mit Hautepithel und dann - etwa 0,2-0,3 mg, mit Kot - etwa 0, 4 mg/Tag. Es ist bekannt, dass Eisen, das im Stuhl verloren geht, Eisen aus abblätterndem Darmepithel, Eisen aus der Galle und exogenes Eisen umfasst, das nicht aus der Nahrung aufgenommen wird. Im Durchschnitt wird angenommen, dass der tägliche Eisenverlust bei Männern und nicht menstruierenden Frauen etwa 1 mg beträgt. Laut verschiedenen Autoren kann der Eisenverlust bei Frauen während einer Menstruation stark variieren - von 2 bis 73 mg.
Häm ist eine prothetische Gruppe vieler Proteine: Hämoglobin, Myoglobin, Cytochrome des mitochondrialen CPE, Cytochrom P 450, das an der mikrosomalen Oxidation beteiligt ist. Die Enzyme Katalase, Peroxidase, Cytochromoxidase enthalten Häm als Coenzym.
Alle Zellen des Körpers haben hämhaltige Proteine, daher findet in allen Zellen eine Häm-Synthese statt, mit Ausnahme von Erythrozyten, die bekanntlich kein Protein-Synthesesystem besitzen.
Beim Abbau von Häm wird in den Zellen des RES der Gallenfarbstoff Bilirubin gebildet. Der weitere Abbau von Bilirubin in Leber, Darm und Niere führt zur Bildung der Endprodukte des Hämabbaus, Stercobilin bzw. Urobilin, die in Kot bzw. Urin enthalten sind. Das beim Abbau von Häm freigesetzte Eisen wird wieder zur Synthese eisenhaltiger Proteine verwendet.
I. STRUKTUR UND BIOSYNTHESE VON HEMA a. Häm-Struktur
Häm besteht aus einem Eisen(II)-Ion und Porphyrin (Abbildung 13-1). Die Struktur der Porphyrine basiert auf Porphin. Porphin besteht aus vier Pyrrolringen, die durch Methenbrücken verbunden sind (Abb. 13-1). Abhängig von der Struktur der Substituenten in Pyrrolringen werden verschiedene Arten von Porphyrinen unterschieden: Protoporphyrine, Ätioporphyrine, Mesoporphyrine und Coproporphyrine. Protoporphyrine sind Vorläufer aller anderen Porphyrine.
Die Häme verschiedener Proteine können enthalten verschiedene Typen Porphyrine (siehe Abschnitt 6). Das Hämoglobin-Häm enthält Protoporphyrin IX, das 4 Methyl-, 2 Vinylreste und 2 Propionsäurereste aufweist. Eisen im Häm befindet sich in einem reduzierten Zustand (Fe+2) und ist durch zwei kovalente und zwei koordinative Bindungen mit den Stickstoffatomen der Pyrrolringe verbunden. Wenn Eisen oxidiert wird, wird Häm umgewandelt
in Hämatin (Fe 3 +). Die größte Menge an Häm ist in mit Hämoglobin gefüllten Erythrozyten, Muskelzellen mit Myoglobin und Leberzellen aufgrund des hohen Gehalts an Cytochrom P 450 enthalten.
B. Häm-Biosynthese
Häm wird in allen Geweben synthetisiert, jedoch mit der höchsten Rate im Knochenmark und in der Leber (Abb. 13-2). Im Knochenmark ist Häm für die Synthese von Hämoglobin in Retikulozyten, in Hepatozyten - für die Bildung von Cytochrom P 450 - notwendig.
Die erste Reaktion der Hämsynthese, die Bildung von 5-Aminolävulinsäure aus Glycin und Succinyl-CoA (Abb. 13-3), findet in der mitochondrialen Matrix statt, wo eines der Substrate dieser Reaktion, Succinyl-CoA, gebildet wird die TCA. Diese Reaktion wird durch das Pyridoxal-abhängige Enzym 5-Aminolävulinat-Synthase katalysiert.
Aus den Mitochondrien gelangt 5-Aminolävulinsäure in das Zytoplasma. Im Zytoplasma finden Zwischenstufen der Hämsynthese statt: die Verbindung von 2 Molekülen 5-Aminolävulinsäure zu einem Molekül Porphobilinogen (Abb. 13-4), Desaminierung von Porphobilinogen unter Bildung von Hydroxymethylbilan, enzymatische Umwandlung von Hydroxymethylbilan zu a Molekül Uroporphobilinogen III, Decarboxylierung des letzteren unter Bildung von Coproporphyrinogen III. Hydroxymethylbilan kann auch nichtenzymatisch zu Uroporphyrinogen I umgewandelt werden, das zu Coproporphyrinogen I decarboxyliert wird. Aus dem Zytoplasma gelangt Coproporphyrinogen III wieder in die Mitochondrien, wo die Endreaktionen der Hämsynthese stattfinden. Als Ergebnis von zwei aufeinanderfolgenden oxidativen Reaktionen wird Coproporphyrinogen III in Protoporphyrinogen IX und Protoporphyrinogen IX - in Protoporphyrin IX umgewandelt. Das Enzym Ferrochelatase wandelt Eisen(II) an Protoporphyrin IX an und wandelt es in Häm um (Abb. 13-2). Das eisenspeichernde Protein Ferritin dient als Eisenquelle für die Hämsynthese. Syn-
Reis. 13-1. Die Struktur von Porphin (A), Protoporphyrin IX (B) und Hämoglobin-Häm (C). Porphin ist eine zyklische Struktur, die aus vier Pyrrolringen besteht, die durch Methenbrücken verbunden sind. Proto-Porphyrin IX besitzt vier Methyl-, zwei Vinylreste und zwei Propionsäurereste. Im Hämoglobin-Häm bildet Fe 2+ zwei kovalente und zwei koordinative Bindungen mit den Stickstoffatomen der Pyrrolringe von Protoporphyrin IX.
Das getestete Häm bildet zusammen mit den α- und β-Polypeptidketten des Globins Hämoglobin. Häm reguliert die Globinsynthese: Wenn die Hämsyntheserate abnimmt, wird die Globinsynthese in Retikulozyten gehemmt.
B. REGULIERUNG DER HEMA-BIOSYNTHESE
Die regulatorische Reaktion der Hämsynthese wird durch das Pyridoxal-abhängige Enzym 5-Aminolävulinat-Synthase katalysiert. Die Reaktionsgeschwindigkeit wird allosterisch und auf der Ebene der Translation des Enzyms reguliert.
Häm ist ein allosterischer Inhibitor und Corepressor der 5-Aminolävulinat-Synthase-Synthese (Abb. 13-5).
In Retikulozyten reguliert die Synthese dieses Enzyms im Stadium der Translation das Eisen. An der Initiationsstelle der für das Enzym kodierenden mRNA befindet sich eine Nukleotidsequenz, die eine Haarnadelschleife bildet, die als eisensensitives Element (aus dem Englischen) bezeichnet wird. auf Eisen reagierendes Element, IRE) (Abbildung 13-6).
Bei hohen Eisenkonzentrationen in Zellen bildet es einen Komplex mit den Cysteinresten des regulatorischen eisenbindenden Proteins. Die Wechselwirkung von Eisen mit dem regulatorischen eisenbindenden Protein verursacht eine Abnahme der Affinität dieses Proteins für das IRE-Element der mRNA, die für 5-Aminolävulinat-Synthase kodiert, und die Fortsetzung der Translation (Fig. 13-6, A). Bei niedrigen Eisenkonzentrationen eisenbindend

Reis. 13-2. Häm-Synthese. Enzyme sind durch die Zahlen im Diagramm gekennzeichnet: 1 - 5-Aminolävulinat-Synthase; 2 - 5-Aminolvulinat-Dehydratase; 3 - Porphobilinogen-Deaminase; 4 - Uroporphyrinogen III-Cosyntase; 5 - Uroporphyrin-Gendecarboxylase; 6 - Coproporphyrinogen III-Oxidase; 7 - Protoporphyrinogenoxidase; 8 - Ferrochelatase. Die Buchstaben bezeichnen die Substituenten in den Pyrrolringen: M – Methyl, B – Vinyl, P – Propionsäurereste, A – Acetyl, PF – Pyridoxalphosphat. Der Eisenspender ist Ferritin, das Eisen in den Zellen speichert.

Reis. 13-3. Die Reaktion der Bildung von 5-Aminolävulinsäure.
das Protein bindet an das eisensensitive Element, das sich am 5'-untranslatierten Ende der mRNA befindet, und die Translation der 5-Aminolävulinat-Synthase wird gehemmt (Abb. 13-6, B).
Auch die 5-Aminolävulinat-Dehydratase wird durch Häm allosterisch gehemmt, da die Aktivität dieses Enzyms jedoch fast 80-mal höher ist als die der 5-Aminolävulinat-Synthase, ist dies physiologisch von geringer Bedeutung.

Reis. 13-4. Porphobilinogen-Bildungsreaktion.

Reis. 13-5. Regulation der Häm- und Hämoglobinsynthese. Häm hemmt nach dem Prinzip der negativen Rückkopplung die 5-Aminolävulinat-Synthase und die 5-Aminolävulinat-Dehydratase und ist ein Translationsinduktor α- und β -Ketten von Hämoglobin.

Reis. 13-6. Regulation der Synthese von Aminolävulinat-Synthase. A – Bei einer hohen Eisenkonzentration in Retikulozyten bindet es an ein eisenbindendes Protein und verringert die Affinität dieses Proteins für ein eisensensitives Element (IRE) der Messenger-RNA, die für 5-Aminolävulinat-Synthase kodiert. Proteinfaktoren Translationsinitiation bindet an mRNA und initiiert die Translation der 5-Aminolävulinat-Synthase. B - Bei einem niedrigen Eisengehalt in Retikulozyten hat das eisenbindende Protein eine hohe Affinität für IRE und interagiert mit diesem. Translkönnen nicht an mRNA binden und die Translation stoppt.
Pyridoxalphosphatmangel und Medikamente, die seine strukturellen Analoga sind, reduzieren die Aktivität der 5-Aminolävulinat-Synthase.
D. STÖRUNGEN DER HEMA-BIOSYNTHESE. PORPHIREN
Erbliche und erworbene Störungen der Hämsynthese, die mit einer Erhöhung des Gehalts an Porphyrinogenen sowie deren Oxidationsprodukten in Gewebe und Blut und ihrem Auftreten im Urin einhergehen, werden als Porphyrien bezeichnet ("Porphyrin" bedeutet auf Griechisch Purpur). Der Urin der Patienten ist rot.
Hereditäre Porphyrien werden durch genetische Defekte in den beteiligten Enzymen verursacht
bei der Hämsynthese, mit Ausnahme der 5-Aminolevulin-Synthase. Bei diesen Erkrankungen wird eine Abnahme der Hämbildung festgestellt. Da Häm ein allosterischer Inhibitor der 5-Aminolävulinat-Synthase ist, nimmt die Aktivität dieses Enzyms zu, was zur Anhäufung von Zwischenprodukten der Häm-Synthese führt - 5-Aminolävulinsäure und Porphyrinogene.
Je nach Hauptlokalisation des pathologischen Prozesses werden hepatische und erythropoetische hereditäre Porphyrien unterschieden. Erythropoetische Porphyrien werden von der Anhäufung von Porphyrinen in Normoblasten und Erythrozyten und hepatischen - in Hepatozyten - begleitet.
Bei schweren Formen der Porphyrie werden neuropsychiatrische Störungen, Funktionsstörungen des RES und Hautschäden beobachtet. Porphyrinogene sind nicht gefärbt und fluoreszieren nicht, werden aber im Licht leicht in Porphyrine umgewandelt. Letztere zeigen eine intensive rote Fluoreszenz in ultravioletten Strahlen. In der der Sonne ausgesetzten Haut geht Sauerstoff durch Wechselwirkung mit Porphyrinen in einen Singulett-Zustand über. Singulett-Sauerstoff bewirkt eine Beschleunigung der Lipidperoxidation von Zellmembranen und eine Zerstörung von Zellen, daher werden Porphyrien oft von Photosensibilisierung und Ulzeration offener Hautbereiche begleitet. Neuropsychiatrische Störungen bei Porphyrien sind damit verbunden, dass 5-Aminolävulinat und Porphyrinogene Neurotoxine sind.
Bei leichten Formen der erblichen Porphyrie kann die Krankheit manchmal asymptomatisch sein, aber die Einnahme von Medikamenten, die die Synthese von 5-Aminolävulinat-Synthase induzieren, kann eine Verschlimmerung der Krankheit verursachen. Induktoren der Synthese von 5-Aminolävulinat-Synthase sind bekannte Arzneimittel wie Sulfanylamide, Barbiturate, Diclofenac, Voltaren, Steroide, Gestagene. In einigen Fällen treten die Krankheitssymptome erst in der Pubertät auf, wenn eine erhöhte Bildung von β-Steroiden die Synthese von 5-Aminolävulinat-Synthase induziert. Porphyrien werden auch bei Vergiftungen mit Bleisalzen beobachtet, da Blei die 5-Aminolävulinat-Dehydratase und Ferrochelatase hemmt. Einige halogenierte Herbizide und Insektizide sind Induktoren der Synthese von 5-Aminolävulinat-Synthase, so dass ihre Einnahme von Porphyrie-Symptomen begleitet wird.
II. EISENWECHSEL
Der Körper eines Erwachsenen enthält 3-4 g Eisen, davon nur etwa 3,5 mg
ist im Blutplasma. Hämoglobin hat ungefähr 68 % des Eisens des gesamten Körpers, Ferritin - 27 %, Myoglobin - 4 %, Transferrin - 0,1 % Alle eisenhaltigen Enzyme machen nur 0,6 % des Eisens im Körper aus. Die Eisenquellen bei der Biosynthese eisenhaltiger Proteine sind Nahrungseisen und Eisen, das beim ständigen Abbau von Erythrozyten in den Zellen der Leber und Milz freigesetzt wird.
In einem neutralen oder alkalischen Medium befindet sich Eisen in einem oxidierten Zustand - Fe 3+ und bildet große, leicht aggregierende Komplexe mit OH -, anderen Anionen und Wasser. Bei niedrigen pH-Werten wird Eisen reduziert und dissoziiert leicht. Der Prozess der Reduktion und Oxidation von Eisen sorgt für seine Umverteilung zwischen Makromolekülen im Körper. Eisenionen haben eine hohe Affinität zu vielen Verbindungen und bilden mit ihnen Chelatkomplexe, die die Eigenschaften und Funktionen dieser Verbindungen verändern; daher wird der Transport und die Ablagerung von Eisen im Körper durch spezielle Proteine bewältigt. In Zellen lagert Eisen das Protein Ferritin ab, im Blut wird es durch das Protein Transferrin transportiert.
A. Eisenaufnahme im Darm
In Lebensmitteln liegt Eisen hauptsächlich in oxidiertem Zustand (Fe 3+) vor und ist Bestandteil von Proteinen oder Salzen organischer Säuren. Die Freisetzung von Eisen aus Salzen organischer Säuren wird durch das saure Milieu des Magensaftes erleichtert. Die größte Menge an Eisen wird im Zwölffingerdarm aufgenommen. Die in der Nahrung enthaltene Ascorbinsäure stellt Eisen wieder her und verbessert seine Aufnahme, da nur Fe 2+ in die Zellen der Darmschleimhaut gelangt. Die tägliche Nahrungsmenge enthält normalerweise 15-20 mg Eisen, von denen nur etwa 10 % resorbiert werden. Der Körper eines Erwachsenen verliert etwa 1 mg Eisen pro Tag.
Die Eisenmenge, die in die Zellen der Darmschleimhaut aufgenommen wird, übersteigt in der Regel den Bedarf des Körpers. Der Eintritt von Eisen aus Enterozyten in das Blut hängt von der Syntheserate des Proteins Apoferritin in ihnen ab. Apoferritin „fängt“ Eisen in Enterozyten und wird zu Ferritin, das in Enterozyten verbleibt. Auf diese Weise werden Erlöse reduziert.
Übertragung von Eisen in Blutkapillaren aus Darmzellen. Bei niedrigem Eisenbedarf erhöht sich die Syntheserate von Apoferritin (siehe unten „Regulierung des Eiseneintrags in die Zellen“). Die ständige Abschuppung von Schleimhautzellen in das Darmlumen befreit den Körper von überschüssigem Eisen. Bei Eisenmangel im Körper wird Apoferritin in Enterozyten fast nicht synthetisiert. Eisen, das von Enterozyten ins Blut gelangt, transportiert das Blutplasmaprotein Transferrin (Abb. 13-7).
B. TRANSPORT VON EISEN IM BLUTPLASMA UND SEINE EMPFANGEN IN DEN ZELLEN
Im Blutplasma transportiert Eisen das Protein Transferrin. Transferrin ist ein Glykoprotein, das in der Leber synthetisiert wird und nur oxidiertes Eisen (Fe 3+) bindet. Eisen, das in den Blutkreislauf gelangt, wird durch ein Enzym namens Ferroxidase oxidiert, das als kupferhaltiges Plasmaprotein Ceruloplasmin bekannt ist. Ein Transferrinmolekül kann ein oder zwei Fe 3+ -Ionen binden, jedoch gleichzeitig mit dem CO 3 2- -Anion, um einen Transferrin-2-Komplex (Fe 3+ -CO 3 2-) zu bilden. Normalerweise ist Bluttransferrin zu etwa 33 % mit Eisen gesättigt.
Transferrin interagiert mit spezifischen Zellmembranrezeptoren.
Durch diese Wechselwirkung wird im Zytosol der Zelle ein Ca 2+ -Calmodulin-PKC-Komplex gebildet, der den Trans-Ferrin-Rezeptor phosphoryliert und die Bildung eines Endosoms bewirkt. Die ATP-abhängige Protonenpumpe, die sich in der Endosomenmembran befindet, erzeugt eine saure Umgebung innerhalb des Endosoms. In der sauren Umgebung des Endosoms wird Eisen aus Transferrin freigesetzt. Danach kehrt der Rezeptor-Apotransferrin-Komplex an die Oberfläche der Zellplasmamembran zurück. Bei einem neutralen pH-Wert der extrazellulären Flüssigkeit ändert Apotransferrin seine Konformation, trennt sich vom Rezeptor, dringt in das Blutplasma ein und wird in der Lage, Eisenionen erneut zu binden und einen neuen Zyklus seines Transports in die Zelle einzuleiten. Eisen wird in der Zelle für die Synthese eisenhaltiger Proteine verwendet oder im Protein Ferritin abgelagert.
Ferritin ist ein oligomeres Protein mit einem Molekulargewicht von 500 kDa. Es besteht aus schweren (21 kD) und leichten (19 kD) Polypeptidketten mit 24 Protomeren. Der unterschiedliche Satz von Protomeren im Ferritin-Oligomer bestimmt die Bildung mehrerer Isoformen dieses Proteins in verschiedenen Geweben. Ferritin ist eine Hohlkugel, die bis zu 4.500 Eisen(III)-Ionen enthalten kann, typischerweise jedoch weniger als 3.000. Schwere Ketten

Reis. 13-7. Der Eintritt von exogenem Eisen in das Gewebe. In der Darmhöhle wird Eisen aus Proteinen und Salzen organischer Säuren in der Nahrung freigesetzt. Die Aufnahme von Eisen wird durch Ascorbinsäure erleichtert, die Eisen reduziert. In den Zellen der Darmschleimhaut verbindet sich überschüssiges Eisen mit dem Protein Apoferritin zu Ferritin, während Ferritin Fe2+ zu Fe3+ oxidiert. Der Eintritt von Eisen aus den Zellen der Darmschleimhaut in das Blut wird von der Oxidation von Eisen durch das Serumenzym Ferroxidase begleitet. Im Blut transportiert Fe3+ das Serumprotein Transferrin. In Geweben wird Fe2+ zur Synthese eisenhaltiger Proteine verwendet oder in Ferritin abgelagert.
Ferritin oxidiert Fe 2+ zu Fe 3+. Eisen in Form von Hydroxidphosphat befindet sich im Zentrum der Kugel, deren Hülle vom Proteinteil des Moleküls gebildet wird. Es tritt ein und wird durch die Kanäle, die die Proteinhülle von Apoferritin durchdringen, nach außen abgegeben, aber Eisen kann auch im Proteinteil des Ferritinmoleküls abgelagert werden. Ferritin kommt in fast allen Geweben vor, die größte Menge jedoch in Leber, Milz und Knochenmark. Ein unbedeutender Teil des Ferritins wird aus den Geweben in das Blutplasma ausgeschieden. Da die Aufnahme von Ferritin im Blut proportional zu seinem Gehalt im Gewebe ist, ist die Ferritinkonzentration im Blut ein wichtiger diagnostischer Indikator für Eisenvorräte im Körper bei Eisenmangelanämie. Der Eisenstoffwechsel im Körper ist in Abb. 13-8.
B. REGELUNG DER EISENZUFUHR IN ZELLEN
Der Eisengehalt in Zellen wird durch das Verhältnis seiner Aufnahme-, Verbrauchs- und Ablagerungsraten bestimmt und wird durch zwei molekulare Mechanismen gesteuert. Die Geschwindigkeit des Eiseneintrags in nicht-erythroide Zellen hängt von der Anzahl der Transferrin-Rezeptorproteine in ihrer Membran ab. Überschüssiges Eisen in den Zellen lagert Ferritin ab. Die Synthese von Apoferrithin- und Transferrinrezeptoren wird auf der Ebene der Translation dieser Proteine reguliert und hängt vom Eisengehalt in der Zelle ab.
Am untranslatierten 3"-Ende der Transferrinrezeptor-mRNA und am untranslatierten 5"-Ende der Apoferritin-mRNA befinden sich Haarnadelschleifen – eisensensitive IRE-Elemente (Abb. 13-9 und 13-10). Darüber hinaus ist die Transferrezeptor-mRNA

Reis. 13-8. Eisenstoffwechsel im Körper.

Reis. 13-9. Regulation der Apoferritin-Synthese. A - Bei einer Abnahme des Eisengehalts in der Zelle hat das eisenbindende Protein eine hohe Affinität zu IRE und interagiert mit diesem. Dies verhindert die Anheftung von Proteian die mRNA, die für Apoferritin kodiert, und die Synthese von Apoferritin stoppt; B - bei einer Erhöhung des Eisengehalts in der Zelle interagiert es mit dem eisenbindenden Protein, wodurch die Affinität dieses Proteins für IRE abnimmt. Translbinden an mRNA, die für Apoferritin kodiert, und initiieren die Translation von Apoferritin.
rina hat 5 Schleifen, während Apoferritin-mRNA nur 1 hat.
Diese Regionen der mRNA können mit dem regulatorischen IRE-bindenden Protein interagieren. Bei niedrigen Eisenkonzentrationen in der Zelle bindet das IRE-bindende Protein an das IRE der Apoferritin-mRNA und verhindert die Bindung von Proteintranslations-Initiationsfaktoren (Abb. 13-9, A). Dadurch sinken die Translationsrate von Apoferritin und sein Gehalt in der Zelle. Gleichzeitig bindet das IRE-bindende Protein bei niedrigen Eisenkonzentrationen in der Zelle an das eisensensitive Element der Transferrinrezeptor-mRNA und verhindert dessen Zerstörung durch das RNAase-Enzym (Abb. 13-10, A). Es bewirkt eine Erhöhung der Anzahl der Rezeptoren
Transferrin und beschleunigen den Eintritt von Eisen in die Zellen.
Bei einer Erhöhung des Eisengehalts in der Zelle werden durch seine Wechselwirkung mit dem IRE-bindenden Protein die SH-Gruppen des aktiven Zentrums dieses Proteins oxidiert und die Affinität zu den eisensensitiven Elementen der mRNA nimmt ab. Dies hat zwei Konsequenzen:
Zunächst wird die Translation von Apoferritin beschleunigt (Abb. 13-9, B);
Zweitens setzt das IRE-bindende Protein die Haarnadelschleifen der Transferrinrezeptor-mRNA frei und wird durch das RNAase-Enzym zerstört, wodurch die Syntheserate der Transferrinrezeptoren abnimmt (Abb. 1310, B). Beschleunigung der Synthese von Apoferritin und Hemmung der Synthese von trans-

Reis. 13-10. Regulation der Transferrinrezeptorsynthese. A – Bei einem niedrigen Eisengehalt in der Zelle hat das eisensensitive Protein eine hohe Affinität für das IRE der mRNA, die das Trans-Ferrin-Rezeptorprotein kodiert. Die Anlagerung eines eisenbindenden Proteins an die mRNA IRE verhindert deren Abbau durch RNAase und die Synthese des Transferrinrezeptorproteins geht weiter; B - Bei einem hohen Eisengehalt in der Zelle nimmt die Affinität des eisenbindenden Proteins für IRE ab und die mRNA wird für die Wirkung der RNAase verfügbar, die sie hydrolysiert. Die Zerstörung von mRNA führt zu einer Abnahme der Synthese des Transferrinrezeptorproteins.
Ferrin bewirken eine Verringerung des Gehalts
Eisen in der Zelle. Im Allgemeinen regulieren diese Mechanismen den Eisengehalt in Zellen und seine Verwendung für die Synthese eisenhaltiger Proteine.
D. STÖRUNGEN DES EISENSTOFFWECHSELS
Eine Eisenmangelanämie kann bei wiederkehrenden Blutungen, Schwangerschaft, häufigen Geburten, Geschwüren und Tumoren auftreten
Magen-Darm-Trakt, nach Operationen am Magen-Darm-Trakt. Bei einer Eisenmangelanämie nehmen die Größe der Erythrozyten und ihre Pigmentierung (hypochrome Erythrozyten kleiner Größe) ab. In Erythrozyten nimmt der Hämoglobingehalt ab, die Sättigung von Transferrin mit Eisen nimmt ab und die Ferritinkonzentration im Gewebe nimmt ab. Der Grund für diese Veränderungen ist der Eisenmangel im Körper, wodurch die Synthese von Häm und Ferritin in nicht-erythroiden Geweben und Hämoglobin in erythroiden Zellen abnimmt.
Hämochromatose. Wenn die Eisenmenge in den Zellen das Volumen des Ferritin-Depots überschreitet, wird Eisen im Proteinteil des Ferritin-Moleküls abgelagert. Durch die Bildung solcher amorpher Ablagerungen von überschüssigem Eisen wird Ferritin in Hämosiderin umgewandelt. Hämosiderin ist schwer wasserlöslich und enthält bis zu 37 % Eisen. Die Ansammlung von Hämosiderin-Granulat in Leber, Bauchspeicheldrüse, Milz führt zu einer Schädigung dieser Organe - Hämochromatose. Hämochromatose kann durch eine erblich erhöhte Eisenaufnahme im Darm verursacht werden, während der Eisengehalt im Körper des Patienten 100 g erreichen kann Diese Krankheit wird autosomal-rezessiv vererbt, und etwa 0,5% der Kaukasier sind homozygot für das Hämochromatose-Gen . Die Anreicherung von Hämosiderin in der Bauchspeicheldrüse führt zur Zerstörung von β-Zellen der Langerhansschen Inseln und in der Folge zu Diabetes mellitus. Die Ablagerung von Hämosiderin in Hepatozyten verursacht eine Leberzirrhose und in Myokardiozyten - Herzinsuffizienz. Patienten mit hereditärer Hämochromatose werden je nach Schweregrad des Patienten mit regelmäßigem Aderlass, wöchentlich oder einmal monatlich, behandelt. Häufige Bluttransfusionen können zu Hämochromatose führen, in diesen Fällen werden die Patienten mit eisenbindenden Medikamenten behandelt.
III. HÄMOGLOBIN-KATABOLISMUS
Rote Blutkörperchen haben eine kurze Lebensdauer (ungefähr 120 Tage). Unter physiologischen Bedingungen werden im Körper eines Erwachsenen etwa 1-2x10 und Erythrozyten pro Tag zerstört. Ihr Katabolismus findet hauptsächlich in den retikuloendothelialen Zellen der Milz, der Lymphknoten, des Knochenmarks und der Leber statt. Mit der Alterung der Erythrozyten nimmt der Gehalt an Sialinsäuren in der Zusammensetzung der Glykoproteine ab Plasma Membran... Die veränderten Kohlenhydratbestandteile der Glykoproteine der Erythrozytenmembranen binden an die Rezeptoren der RES-Zellen und die Erythrozyten werden durch Endozytose in diese „eingetaucht“. Der Abbau von Erythrozyten in diesen Zellen beginnt mit dem Abbau von Hämoglobin in Häm und Globin und anschließender Hydrolyse des Proteinanteils des Hämoglobins durch lysosomale Enzyme.
A. HEMA-KATABOLISMUS
Die erste Reaktion des Hämkatabolismus erfolgt unter Beteiligung von NADPH-abhängigen Enzymkomplex Hämoxygenase. Das Enzymsystem ist in der ER-Membran lokalisiert, im Bereich der Elektronentransportketten der mikrosomalen Oxidation. Das Enzym katalysiert die Spaltung der Bindung zwischen zwei Pyrrolringen mit Vinylresten und öffnet so die Ringstruktur (Abb. 13-11). Im Laufe der Reaktion bildet sich eine lineare Tetrapyr-Walze - biliverdin(ein gelbes Pigment) und Kohlenmonoxid (CO), das sich vom Kohlenstoff der Methenylgruppe ableitet. Häm induziert die Transkription des Häm-Oxygenase-Gens, das für Häm absolut spezifisch ist.
Beim Abbau von Häm freigesetzte Eisenionen können zur Synthese neuer Hämoglobinmoleküle oder zur Synthese anderer eisenhaltiger Proteine verwendet werden. Biliverdin wird durch ein NADPH-abhängiges Enzym zu Bilirubin reduziert Biliverdinreduktase. Bilirubin wird nicht nur beim Abbau von Hämoglobin gebildet, sondern auch beim Abbau anderer hämhaltiger Proteine wie Cytochrome und Myoglobin. Beim Zerfall von 1 g Hämoglobin werden 35 mg Bilirubin und bei einem Erwachsenen etwa 250-350 mg Bilirubin pro Tag gebildet. Der weitere Metabolismus von Bilirubin erfolgt in der Leber.
B. STOFFWECHSEL VON BILIRUBIN
In den Zellen des RES (Milz und Knochenmark) gebildetes Bilirubin ist schwer wasserlöslich und wird im Komplex mit dem Blutplasmaprotein Albumin durch das Blut transportiert. Diese Form von Bilirubin wird als unkonjugiertes Bilirubin bezeichnet. Jedes Albuminmolekül bindet 2 (oder sogar 3) Bilirubinmoleküle, von denen eines stärker (höhere Affinität) an das Protein gebunden ist als die anderen. Wenn sich der pH-Wert des Blutes auf die saure Seite verschiebt (eine Erhöhung der Konzentration von Ketonkörpern, Laktat), nimmt die Ladung, die Konformation des Albumins und die Affinität für Bilirubin ab. Daher ist an Albumin gebundenes Bilirubin fragil, kann von den Bindungsstellen verdrängt werden und bildet Komplexe mit dem extrazellulären Matrixkollagen und Membranlipiden. Eine Reihe von Arzneimitteln konkurrieren mit Bilirubin um

Reis. 13-11. Häm Aufschlüsselung. M - (-CH 3) - Methylgruppe; B – (-CH = CH 2 ) – Vinylgruppe; P – (-CH 2 -CH 2 -COOH) – Propionsäurerest. Während der Reaktion wird eine Methylgruppe in Kohlenmonoxid umgewandelt und damit die Ringstruktur geöffnet. Das gebildete Biliverdin wird unter der Wirkung der Biliverdin-Reduktase in Bilirubin umgewandelt.
hochaffines, hochaffines Zentrum von Albumin.
Aufnahme von Bilirubin durch Parenchymzellen der Leber
Der Albumin-Bilirubin-Komplex, der mit dem Blutstrom an die Leber abgegeben wird, an der Oberfläche
der Plasmamembran der Hepatozyten dissoziiert. Das freigesetzte Bilirubin bildet einen temporären Komplex mit Plasmamembranlipiden. Die erleichterte Diffusion von Bilirubin in die Hepatozyten erfolgt durch zwei Arten von Trägerproteinen: Ligandin (es transportiert den Großteil der
Rubin) und Protein Z. Die Aktivität der Absorption von Bilirubin durch Hepatozyten hängt von der Geschwindigkeit seines Stoffwechsels in der Zelle ab.
Ligandin und Protein Z kommen auch in den Zellen der Niere und des Darms vor und können daher bei unzureichender Leberfunktion die Abschwächung der Entgiftungsprozesse in diesem Organ ausgleichen.
Konjugation von Bilirubin im glatten ER
Im glatten ER von Hepatozyten sind Reste von Glucuronsäure an Bilirubin-Konjugationsreaktionen gebunden. Bilirubin hat 2 Carboxylgruppen, daher kann es sich mit 2 Molekülen Glucuronsäure verbinden und ein in Wasser leicht lösliches Konjugat bilden - Bilirubindiglucuronid (konjugiertes oder direktes Bilirubin) (Abb. 13-12).
Der Donor von Glucuronsäure ist UDP-Glucuronat. Spezifische Enzyme, UDP-Glucuronyltransferasen (Uridindiphosphoglucuronyltransferasen) katalysieren die Bildung von Bilirubinmono- und -diglucuroniden (Abb. 13-13). Einige Arzneimittel, beispielsweise Phenobarbital (siehe Abschnitt 12), dienen als Induktoren der Synthese von UDP-Glucuronyltransferasen.
Bilirubinsekretion in die Galle
Die Sekretion von konjugiertem Bilirubin in die Galle erfolgt über einen aktiven Transportmechanismus, d.h. gegen den Konzentrationsgradienten. Der aktive Transport ist wahrscheinlich die geschwindigkeitsbestimmende Stufe des gesamten Prozesses des Bilirubinstoffwechsels in der Leber. Diglucuronid ist normal

Reis. 13-12. Die Struktur von Bilirubindiglucuronid (konjugiertes, "direktes" Bilirubin). Glucuronsäure wird durch eine Etherbindung an zwei Propionsäurereste addiert, um Acylglucuronid zu bilden.
Bilirubin ist die Hauptausscheidungsform von Bilirubin über die Galle, jedoch ist das Vorhandensein einer geringen Menge an Monoglucuronid nicht ausgeschlossen. Der Transport von konjugiertem Bilirubin von der Leber zur Galle wird durch dieselben Medikamente aktiviert, die eine Bilirubin-Konjugation induzieren können. Somit können wir sagen, dass die Konjugationsrate von Bilirubin und der aktive Transport von bilirubierendem Glucuronid von den Hepatozyten zur Galle eng miteinander verbunden sind (Abb. 13-14).
C. KATABOLISMUS VON BILIRUBINDIGLUCURONID
Im Darm werden die aufgenommenen bilirubing Glucuronide durch spezifische bakterielle Enzyme β-Glucuronidasen hydrolysiert, die die Bindung zwischen Bilirubin und dem Glucuronsäurerest hydrolysieren. Kostenlos

Reis. 13-13. Bildung von Bilirubindiglucuronid.

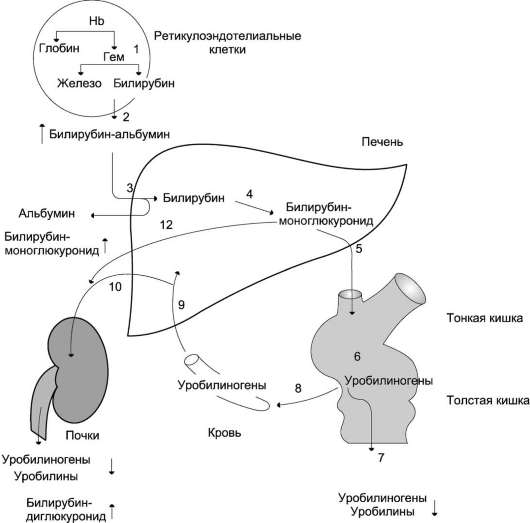
Reis. 13-14. Bilirubin-Urobilinogen-Zyklus in der Leber. 1 - Hb-Katabolismus in retikuloendothelialen Zellen des Knochenmarks, der Milz, der Lymphknoten; 2 - die Bildung der Transportform des Bilirubin-Albumin-Komplexes; 3 - der Fluss von Bilirubin in die Leber; 4 - die Bildung von bilirubing Glucuroniden;
5 - Sekretion von Bilirubin in der Galle in den Darm;
6 - Katabolismus von Bilirubin unter dem Einfluss von Darmbakterien; 7 - Entfernung von Urobilinogenen mit Kot; 8 - Aufnahme von Urobilinogenen in das Blut; 9 - Aufnahme von Urobilinogenen durch die Leber; 10 - der Eintritt eines Teils der Urobilinogene in das Blut und die Ausscheidung durch die Nieren im Urin; 11 - Ein kleiner Teil der Urobilinogene wird in die Galle ausgeschieden.
Das bei dieser Reaktion produzierte Bilirubin wird durch die Wirkung der Darmflora reduziert, um eine Gruppe farbloser Tetrapyrrolverbindungen zu bilden - Urobilinogene(Abb. 13-15).
Im Ileum und Dickdarm wird ein kleiner Teil der Urobilinogene resorbiert und gelangt mit dem Blut der Pfortader in die Leber. Der Hauptteil des Urobilinogens aus der Leber als Bestandteil der Galle wird in den Darm ausgeschieden und mit
Kot aus dem Körper, ein Teil des Urobilinogens aus der Leber gelangt in den Blutkreislauf und wird in Form von Urobilin mit dem Urin ausgeschieden (Abb. 13-14). Normalerweise werden die meisten der im Dickdarm gebildeten farblosen Urobilinogene im Enddarm unter Einwirkung der Darmflora zu einem braunen Pigment oxidiert. urobilin und wird mit Kot entfernt. Die Stuhlfarbe ist auf das Vorhandensein von Urobilin zurückzuführen.

Reis. 13-15. Die Struktur einiger Gallenfarbstoffe. Mesobilinogen ist ein Zwischenprodukt des Bilirubin-Katabolismus im Darm.
NS. Diagnosewert
Bestimmung der Konzentration von Bilirubin in biologische Flüssigkeiten Mensch
Zur Bestimmung des Bilirubingehalts im Blutserum (Plasma) wird derzeit die 1916 von Van der Berg vorgeschlagene Methode zur Bestimmung von Bilirubin im Blutserum auf der Grundlage einer Diazoreaktion verwendet.
V normale Vorraussetzungen die Konzentration des Gesamtbilirubins im Plasma beträgt 0,3-1 mg / dl (1,7-17 µmol / l), 75% des Gesamtbilirubins liegen in unkonjugierter Form (indirektes Bilirubin) vor. In der Klinik wird co-konjugiertes Bilirubin als direkt bezeichnet, da es wasserlöslich ist und schnell mit einem Diazoreagens interagieren kann, wodurch eine rosafarbene Verbindung gebildet wird - dies ist die direkte Reaktion von Van der Berg. Unkonjugiertes Bilirubin ist hydrophob, daher ist es im Blutplasma in einem Komplex mit Albumin enthalten und reagiert erst mit einem Diazoreaktiven, wenn ein organisches Lösungsmittel wie Ethanol zugegeben wird, das Albumin ausfällt. Unkonjugiertes Bilirubin, das erst nach Proteinfällung mit dem Azofarbstoff wechselwirkt, wird als indirektes Bilirubin bezeichnet.
Bei Patienten mit hepatozellulärer Pathologie, begleitet von einem verlängerten Anstieg der
Konjugierte Bilirubinkonzentrationen im Blut weisen auf eine dritte Form des Plasmabilirubins hin, bei der Bilirubin kovalent an Albumin gebunden ist und daher nicht auf übliche Weise getrennt werden kann. In einigen Fällen können bis zu 90 % des gesamten Bilirubingehalts im Blut in dieser Form vorliegen.
A. JELTUKHI
Hyperbilirubinämie kann durch eine Erhöhung der Bilirubinproduktion über die Fähigkeit der Leber hinaus, es auszuscheiden, oder durch eine Schädigung der Leber verursacht werden, was zu einer verminderten Sekretion von Bilirubin in die Galle in normalen Mengen führt. Hyperbilirubinämie wird auch mit Blockierung der Gallenwege der Leber festgestellt.
In allen Fällen steigt der Gehalt an Gesamtbilirubin im Blut an. Wenn eine bestimmte Konzentration erreicht ist, diffundiert es in das Gewebe und färbt es gelb. Eine Gelbfärbung des Gewebes aufgrund der Ablagerung von Bilirubin wird als Gelbsucht bezeichnet. Klinisch kann Gelbsucht erst auftreten, wenn die Konzentration von Bilirubin im Blutplasma die obere Normgrenze um mehr als das 2,5-fache überschreitet, d. steigt nicht über 50 μmol / L an.
1. Hämolytische (suprahepatische) Gelbsucht
Es ist bekannt, dass die Fähigkeit der Leber, Glucuronide zu bilden und in die Galle auszuscheiden, 3-4 mal höher ist als ihre Bildung unter physiologischen Bedingungen. Hämolytische (suprahepatische) Gelbsucht ist das Ergebnis einer intensiven Hämolyse von Erythrozyten. Es wird durch die übermäßige Produktion von Bilirubin verursacht, die über
die Fähigkeit der Leber, es auszuscheiden. Hämolytische Gelbsucht entsteht, wenn die Reservekapazitäten der Leber erschöpft sind. Die Hauptursache des suprahepatischen Ikterus sind erbliche oder erworbene hämolytische Anämien. Bei hämolytischen Anämien durch Sepsis, Strahlenkrankheit, Mangel an Glucose-6-Phosphat-Dehydrogenase von Erythrozyten, Thalassämie, Transfusion inkompatibler Blutgruppen, Vergiftung mit Sulfonamiden kann die Menge an Hämoglobin, die aus Erythrozyten pro Tag freigesetzt wird, 45 g . erreichen
(in einer Menge von 6,25 g), was die Bildung von Bilirubin signifikant erhöht. Hyperbilirubinämie bei Patienten mit hämolytischem Ikterus wird durch einen signifikanten Anstieg (103-171 μmol / L) der Konzentration von Albumin-gebundenem unkonjugiertem Bilirubin (indirektes Bilirubin) im Blut verursacht. Die Bildung großer Mengen von Biliru-Binglucuroniden (direktes Bilirubin) in der Leber und der Eintrag in den Darm führt zu einer verstärkten Bildung und Ausscheidung von Urobilinogenen mit Kot und Urin und deren intensiverer Färbung (Abb. 13-16).

Reis. 13-16. Bilirubin-Urobilinogen-Zyklus bei hämolytischer Gelbsucht. 1 - Der Hb-Katabolismus schreitet mit erhöhter Geschwindigkeit voran; 2 - die Konzentration von indirektem Bilirubin im Blut ist etwa 10-mal erhöht; 3 - Albumin wird aus dem Bilirubin-Albumin-Komplex freigesetzt; 4 - die Aktivität der Glucuronidierungsreaktion nimmt zu, ist jedoch niedriger als die Geschwindigkeit der Bilirubinbildung; 5 - die Sekretion von Bilirubin in die Galle ist erhöht; 6, 7, 10 - der erhöhte Gehalt an Urobilinogenen in Kot und Urin verleiht ihnen eine intensivere Farbe; Urobilinogen wird aus dem Darm ins Blut aufgenommen (8) und gelangt über die Pfortader (9) in die Leber.
Eines der Hauptzeichen einer hämolytischen Gelbsucht ist ein Anstieg des unkonjugierten (indirekten) Bilirubins im Blut. Dies macht es leicht, ihn von mechanischem (subhepatischem) und hepatozellulärem (hepatischem) Ikterus zu unterscheiden.
Unkonjugiertes Bilirubin ist giftig. Hydrophobes, lipophiles unkonjugiertes Bilirubin, das sich leicht in Membranlipiden auflöst und dadurch in die Mitochondrien eindringt, entkoppelt dort Atmung und oxidative Phosphorylierung, stört die Proteinsynthese, den Fluss von Kaliumionen durch die Zellmembran und Organellen. Dies wirkt sich negativ auf den Zustand des Zentralnervensystems aus und verursacht bei Patienten eine Reihe charakteristischer neurologischer Symptome.
Gelbsucht bei Neugeborenen
Eine besondere Art der hämolytischen Gelbsucht bei Neugeborenen ist die "physiologische Gelbsucht", die in den ersten Lebenstagen eines Kindes beobachtet wird. Der Grund für die Erhöhung der Konzentration von indirektem Bilirubin im Blut ist eine beschleunigte Hämolyse und eine unzureichende Funktion von Leberproteinen und Enzymen, die für die Absorption, Konjugation und Sekretion von direktem Bilirubin verantwortlich sind. Bei Neugeborenen ist nicht nur die Aktivität der UDP-Glucuronyltransferase verringert, sondern anscheinend ist die Synthese des zweiten Substrats der Konjugationsreaktion von UDP-Glucuronat nicht ausreichend aktiv.
Es ist bekannt, dass UDP-Glucuronyltransferase ein induzierbares Enzym ist (siehe Abschnitt 12). Neugeborene mit physiologischer Gelbsucht erhalten das Medikament Phenobarbital, dessen induzierende Wirkung in Abschnitt 12 beschrieben wurde.
Eine der unangenehmen Komplikationen der "physiologischen Gelbsucht" ist die Bilirubin-Enzephalopathie. Wenn die Konzentration von unkonjugiertem Bilirubin 340 μmol / L überschreitet, passiert es die Blut-Hirn-Schranke des Gehirns und verursacht dessen Schädigung.
2. Hepatozelluläre (hepatische) Gelbsucht
Hepatozellulärer (hepatischer) Ikterus wird durch Schäden an Hepatozyten und Gallenkapillaren verursacht, beispielsweise bei akuten Virusinfektionen, chronischer und toxischer Hepatitis.
Der Grund für die Erhöhung der Bilirubinkonzentration im Blut ist die Niederlage und Nekrose eines Teils der Leberzellen. Es kommt zu einer Verzögerung des Bilirubins in der Leber, die durch eine starke Abschwächung der Stoffwechselprozesse in den betroffenen Hepatozyten erleichtert wird, die ihre Fähigkeit verlieren, verschiedene biochemische und physiologische Funktionen normal auszuführen, insbesondere konjugiertes (direktes) Bilirubin aus Zellen zu übertragen gegen den Konzentrationsgradienten in die Galle. Für den hepatozellulären Ikterus ist charakteristisch, dass in der betroffenen Leberzelle statt der normalerweise vorherrschenden Bilirubin-Diglucuronide hauptsächlich Monoglucuronide gebildet werden.
(Abb. 13-17).
Durch die Zerstörung des Leberparenchyms gelangt das gebildete direkte Bilirubin teilweise in den systemischen Kreislauf, was zu Gelbsucht führt. Auch die Gallenausscheidung ist beeinträchtigt. Es gelangt weniger Bilirubin in den Darm als normal.
Bei hepatozellulärer Gelbsucht steigt die Konzentration sowohl des Gesamtbilirubins als auch seiner beiden Fraktionen - unkonjugiert (indirekt) und konjugiert (direkt) - im Blut.
Da weniger bilirubing Glucuronid in den Darm gelangt, wird auch die Menge an gebildetem Urobilinogen reduziert. Daher ist Kot hypocholisch, d.h. weniger farbig. Urin hingegen hat eine intensivere Farbe, da dort nicht nur Urobiline, sondern auch konjugiertes Bilirubin vorhanden ist, das in Wasser leicht löslich ist und mit dem Urin ausgeschieden wird.
3. Mechanischer oder obstruktiver (subhepatisch-nächtlicher) Gelbsucht
Mechanisch oder obstruktiv (subhepatisch-nächtlich) entwickelt sich Gelbsucht unter Verletzung der Gallensekretion im Zwölffingerdarm. Sie kann durch eine Verstopfung der Gallenwege verursacht werden, z. B. bei Gallensteinen, Tumoren der Bauchspeicheldrüse, der Gallenblase, der Leber, des Zwölffingerdarms, einer chronischen Entzündung der Bauchspeicheldrüse oder einer postoperativen Verengung des Hauptgallengangs (Abb. 13-18) .
Bei vollständiger Blockade des Hauptgallengangs, konjugiertes Bilirubin in der Zusammensetzung
Reis. 13-17. Verletzung des Bilirubin-Urobilinogen-Zyklus bei hepatozellulärer Gelbsucht. In der Leber ist die Reaktionsgeschwindigkeit der Glukuronidierung von Bilirubin reduziert (4), daher steigt die Konzentration von indirektem Bilirubin im Blut; aufgrund einer Verletzung des Leberparenchyms gelangt ein Teil des in der Leber gebildeten bilirubing Glucuronids in das Blut (12) und wird dann mit dem Urin aus dem Körper entfernt (10). Im Urin von Patienten sind Urobiline und bilirubing Glucuronide vorhanden. Die restlichen Zahlen entsprechen den Stadien des Bilirubinstoffwechsels in Abb. 13-16.
Galle gelangt nicht in den Darm, obwohl Hepatozyten sie weiterhin produzieren. Da Bilirubin nicht in den Darm gelangt, gibt es keine Produkte seines Katabolismus Urobilinogene in Urin und Kot. Der Kot ist verfärbt. Da die normalen Ausscheidungswege von Bilirubin blockiert sind, gelangt es ins Blut, wodurch die Konzentration von konjugiertem Bilirubin im Blut der Patienten erhöht wird. Lösliches Bilirubin wird mit dem Urin ausgeschieden, was ihm eine satte orange-braune Farbe verleiht.
B. DIFFERENZDIAGNOSE VON GELB
Bei der Diagnose von Gelbsucht ist zu beachten, dass Gelbsucht jeder Art in der Praxis selten in "reiner" Form festgestellt wird. Eine Kombination der einen oder anderen Art ist häufiger. Bei schwerer hämolytischer Gelbsucht, begleitet von einer Erhöhung der Konzentration von indirektem Bilirubin, leiden also unweigerlich verschiedene Organe, einschließlich der Leber, die Elemente einführen kann

Reis. 13-18. Verletzung des Bilirubin-Urobilinogen-Zyklus bei obstruktiver Gelbsucht. Aufgrund der Blockade der Gallenblase wird bilirubing Glucuronid nicht in die Galle ausgeschieden (5); das Fehlen von Bilirubin im Darm führt zu einer Verfärbung des Kots (6); lösliches bilirubing Glucuronid wird über die Nieren mit dem Urin ausgeschieden (10). Es gibt keine Urobiline im Urin; in der Leber gebildetes Bilirubing-Glucuronid gelangt in den Blutkreislauf (12), wodurch der Gehalt an direktem Bilirubin ansteigt. Die restlichen Zahlen entsprechen den Stadien des Bilirubinstoffwechsels in Abb. 13-16.
parenchymale Gelbsucht, d.h. Anstieg des direkten Bilirubins im Blut und Urin. Parenchymgelbsucht wiederum umfasst in der Regel mechanische Elemente. Bei subhepatischer (mechanischer) Gelbsucht, beispielsweise bei Bauchspeicheldrüsenkopfkrebs, ist eine erhöhte Hämolyse infolge einer Krebsintoxikation und infolgedessen ein Anstieg des direkten und indirekten Bilirubins im Blut unvermeidlich.
Hyperbilirubinämie kann also das Ergebnis eines Überschusses an gebundenem und freiem . sein
Bilirubin. Bei der Diagnose von Gelbsucht ist eine separate Messung ihrer Konzentrationen erforderlich. Wenn die Konzentration von Bilirubin im Plasma<100 мкмоль/л и другие тесты функции печени дают нормальные результаты, возможно предположить, что повышение обусловлено за счёт непрямого билирубина. Чтобы подтвердить это, можно сделать анализ мочи, поскольку при повышении концентрации непрямого билирубина в плазме прямой билирубин в моче отсутствует.
Bei der Differentialdiagnose von Gelbsucht muss der Gehalt an Urobilinogenen im Urin berücksichtigt werden. Normalerweise werden etwa 4 mg Urobilinogene pro Tag mit dem Urin aus dem Körper ausgeschieden. Wird eine erhöhte Menge an Urobilinogenen im Urin ausgeschieden, so ist dies ein Hinweis auf eine ungenügende Leberfunktion, beispielsweise bei hepatischer oder hämolytischer Gelbsucht. Das Vorhandensein von nicht nur Urobilinogenen im Urin, sondern auch von direktem Bilirubin weist auf Leberschäden und eine Verletzung des Gallenflusses in den Darm hin.
B. VERERBTE STÖRUNGEN DES BILIRUBIN-METABOLISMUS
Es sind mehrere Krankheiten bekannt, bei denen Gelbsucht durch erbliche Störungen des Bilirubinstoffwechsels verursacht wird.
Bei etwa 5 % der Bevölkerung wird eine erbliche Gelbsucht diagnostiziert, die durch genetische Störungen in der Struktur von Proteinen und Enzymen verursacht wird, die für den Transport (Einfang) von indirektem Bilirubin zur Leber und dessen Konjugation mit Glucuronsäure verantwortlich sind. Diese Pathologie wird autosomal-dominant vererbt. Im Blut von Patienten ist die Konzentration von indirektem Bilirubin erhöht.
Es gibt 2 Arten von erblicher Gelbsucht, die durch eine Verletzung der Glucuronidierungsreaktion in der Leber verursacht wird - die Bildung von direktem Bilirubin.
Der erste Typ ist durch das vollständige Fehlen von UDP-Glucuronyltransferase gekennzeichnet. Krankheit-
es wird autosomal-rezessiv vererbt. Die Einführung von Phenobarbital, einem Induktor der UDP-Glucuronyltransferase, führt nicht zu einer Senkung des Bilirubinspiegels. Kinder sterben in jungen Jahren an der Entwicklung einer Bilirubin-Enzephalopathie.
Der zweite Typ ist durch eine Abnahme der Aktivität (Mangel) der UDP-Glucuronyl-Transferase gekennzeichnet, Hyperbilirubinämie tritt aufgrund von indirektem Bilirubin auf. Gelbsucht spricht gut auf die Behandlung mit Phenobarbital an.
Eine Störung des aktiven Transports von bilirubierenden Glucuroniden, die in Leberzellen gebildet werden, in die Galle ist charakteristisch für die autosomal-dominant vererbte Gelbsucht. Es manifestiert sich durch Hyperbilirubinämie aufgrund von direktem Bilirubin und Bilirubinurie (direktes Bilirubin wird im Urin bestimmt).
Familiäre neonatale Hyperbilirubinämie ist mit dem Vorhandensein von kompetitiven Inhibitoren der Bilirubin- (Östrogen, freie Fettsäure)-Konjugation in der Muttermilch verbunden. Beim Stillen finden sich im Serum des Kindes Bilirubin-Konjugationshemmer. Diese Hyperbilirubinämie wurde als vorübergehend bezeichnet. Die Hyperbilirubinämie verschwindet, wenn das Kind auf künstliche Ernährung umgestellt wird. Eine refraktäre Hyperbilirubinämie führt zu einer Bilirubin-Enzephalopathie und einem frühen Tod.
Eisenmangelanämie. Die häufigste Ursache für Eisenmangel im Körper ist der Blutverlust, wodurch die Aufnahme von Eisen in den Körper mit der Nahrung im Verhältnis zu seiner Verwertung bei der Bildung roter Blutkörperchen gering wird.
Insbesondere kann eine Eisenmangelanämie verursacht werden durch: Blutungen aus Blutgefäßen, die durch die Bildung von Magen- und Zwölffingerdarmgeschwüren geschädigt wurden, Menstruationsblutverlust. Manchmal überwiegt bei Neugeborenen und Kindern die Verwertung von Eisen für die Erythropoese vor dem Eintritt in den Körper, was zu einer Eisenmangelanämie ohne Blutverlust führt.
Anämie aufgrund chronischer entzündlicher Prozesse. Patienten mit langfristigen (mehr als einen Monat) Erkrankungen, deren Pathogenese überwiegend chronische Entzündungen sind, entwickeln in der Regel eine leichte bis mittelschwere Anämie. Darüber hinaus hängt die Schwere der Anämie direkt mit der Dauer und Schwere des Entzündungsprozesses zusammen. Die am häufigsten zu einer Anämie dieser Art führenden Erkrankungen sind die subakute bakterielle Endokarditis, Osteomyelitis, Lungenabszess, Tuberkulose und Pyelonephritis. Bei Autoimmunerkrankungen werden Autoantikörper-Autoantigen-Immunkomplexe auf der Oberfläche von Zellen des betroffenen Gewebes oder Organs gebildet. Dies führt zur Aktivierung des Komplementsystems entlang des klassischen Weges als auslösender Moment einer Entzündung, die Gewebe und Organe des Patienten schädigt. Daher sind viele der Autoimmunerkrankungen als Erkrankungen anzusehen, die weitgehend durch eine ausgeprägte chronische Entzündung gekennzeichnet sind. Die rheumatoide Arthritis ist die häufigste Autoimmunerkrankung, die aufgrund einer chronischen Entzündung zu Anämie führt.
Eine der Ursachen für Anämie bei Patienten mit bösartigen Neubildungen ist mit einer chronischen Entzündung verbunden.
Zu den unmittelbaren Ursachen einer chronischen entzündungsbedingten Anämie gehören:
1. Hemmung der Bildung von Erythrozyten durch das Knochenmark infolge seiner verlängerten Stimulation mit Zytokinen (koloniestimulierende Faktoren), die von zellulären Effektoren chronischer Entzündungen gebildet und freigesetzt werden.
2. Nichtkompensation der Abnahme der Lebensdauer der Erythrozyten im Blut.
Bei Anämie aufgrund einer chronischen Entzündung ist eine Abnahme des Eisengehalts in Erythroblasten eine Folge einer Verletzung seiner Abgabe an die sich entwickelnden Erythrozyten im Knochenmark. Eisenmangel in Erythrozyten führt zu Hypochromie und Mikrozytose der Erythrozyten. Ein Mangel an Eisen, das für die Hämoglobinsynthese verfügbar ist, führt zu einer Erhöhung des Gehalts an Protoporphyrin in Erythrozyten. Die für die Erythropoese verfügbare Eisenmasse wird trotz seines normalen Gehalts im Körper durch übermäßige systemische Aktivierung mononukleärer Phagozyten sowie durch eine Zunahme ihrer Anzahl (Hyperplasie) reduziert. Als Folge von Hyperplasie und Hyperaktivierung im System der mononukleären Phagozyten kommt es zu einer übermäßigen Aufnahme von Eisen durch aktivierte mononukleäre Zellen mit einer erhöhten Fähigkeit, dieses Mikroelement zu absorbieren. Die erhöhte Fähigkeit mononukleärer Zellen, Eisen aufzunehmen, ist größtenteils auf die hohe Konzentration von Interleukin-1 im zirkulierenden Blut zurückzuführen, die durch chronische Entzündungen ansteigt. Unter dem Einfluss von Interleukin-1, das im Blut zirkuliert und in erhöhter Konzentration in den Interzellularräumen vorliegt, setzen Neutrophile des Gesamtorganismus intensiv Lactoferrin frei.
Dieses Protein bindet freies Eisen, das bei der Zerstörung sterbender roter Blutkörperchen freigesetzt wird, und transportiert es in erhöhten Mengen zu mononuklearen Zellen, die dieses Mikroelement einfangen und zurückhalten. Als Ergebnis entwickelt sich eine mäßige Hemmung der Erythropoese aufgrund einer Abnahme der Verfügbarkeit von Eisen für die Bildung von Erythrozyten.
Vermutlich kann als ein Glied in der Pathogenese von Anämien durch chronische Entzündungen eine übermäßige Zerstörung von Erythrozyten durch Hyperaktivierung und Hyperplasie im System der mononukleären Fresszellen angesehen werden. Dies wird durch die Verkürzung der Lebensdauer fast normaler Erythrozyten belegt, deren pathologische Veränderungen auf einen reduzierten Eisengehalt und eine Erhöhung des Protoporphyringehalts reduziert werden.
Sideroblastische Anämien. Anämien dieser Art sind mit einer gestörten Hämsynthese als Bestandteil des Hämoglobins verbunden. Störungen der Hämoglobinsynthese bei sideroblastischen Anämien charakterisieren die Ansammlung von Eisen in Mitochondrien, die um den Kern abnormer Erythrozyten (Sideroblasten) lokalisiert sind. Diese Zellen werden "beringt" genannt, weil intrazelluläre Eisenablagerungen eine ringförmige Kontur um den Zellkern bilden. Störungen der Hämsynthese bei Patienten mit syroblastischen Anämien führen zu Hypochromie und Mikrozytose.
Es gibt zwei Haupttypen von sideroblastischen Anämien:
1. Die hereditäre sideroblastische Anämie ist eine monogene Erkrankung, deren Übertragung von den Eltern auf den Patienten mit dem X-Chromosom assoziiert oder autosomal-rezessiv vererbt wird. Vermutlich wird die hereditäre sideroblastische Anämie durch einen angeborenen Mangel in der Aktivität des Enzyms Gamma-Aminolävulinsäure-Synthetase (dem Schlüsselenzym der ersten Stufe der Porphyrinsynthese) verursacht. Die Hemmung der Enzymaktivität kann primär oder Folge einer angeborenen Stoffwechselstörung seines essentiellen Cofaktors Pyridoxal-5'-phosphat sein.
2. Erworbene sideroblastische Anämien treten häufiger auf als hereditäre. Erworbene sideroblastische Anämien können durch Nebenwirkungen von Medikamenten (Isoniazid usw.) entstehen. Außerdem können sie idiopathisch sein.
Eine Störung der Eisenverwertung für die Hämbildung bei sideroblastischen Anämien äußert sich in einer Erhöhung des Gehalts seiner Ionen im Blutserum sowie einer Erhöhung der Ferritinkonzentration darin.
Thalassämie ist eine monogene Erkrankung, die auf der Hemmung der Synthese einer der Polymerketten beruht, aus denen das Globinmolekül besteht. Je nach Art der Kette, deren Synthese beim Patienten reduziert ist, wird die Thalassämie in eine von drei Hauptgruppen eingeteilt:
1. Alpha-Thalassämie. Diese Krankheiten werden durch die Deletion (Deletion) des Genoms des Organismus von Alpha-Globin-Genen verursacht. Es gibt vier solcher Gene. Je nachdem, welches Gen durch das Genom verloren geht, reicht die sideroblastische Anämie im Schweregrad von leicht und ohne erkennbare klinische Manifestationen bis hin zu schweren, die den Tod des Fötus im Mutterleib verursachen.
2. Beta-Thalassämien, die durch das Fehlen oder Fehlen des entsprechenden Gens verursacht werden. Wenn ein Gen dysfunktional ist, erfolgt seine Transkription, führt jedoch zur Bildung von abnormaler RNA. Darüber hinaus kann eine Gendysfunktion auch in einer verminderten Produktion von normaler RNA bestehen. Das Genom enthält zwei verschiedene Gene für Beta-Globin. Daher gibt es zwei Arten von Beta-Thalassämien. Bei einer schwereren Form der Beta-Thalassämie (Kuley-Anämie) werden deren Symptome bereits im Kindesalter erkannt. Im Alter von dreißig Jahren tritt in der Regel trotz Bluttransfusion der Tod ein. Bei weniger schwerer Beta-Thalassämie besteht keine Indikation für eine Bluttransfusion und eine Anämie schränkt die Lebenserwartung nicht ein.
Bei der Untersuchung eines Blutausstrichs zeigt sich neben Hypochromie und Mikrozytose bei Patienten mit Thalassämie eine Poikilozytose, dh eine pathologische Variabilität in Form von Erythrozyten.
Hämoglobin wird in den Zellen des Knochenmarks synthetisiert. Alle für die Hämoglobinsynthese notwendigen Bestandteile kommen mit dem Blutkreislauf.
Der Proteinanteil des Moleküls wird wie alle einfachen Proteine aus Aminosäuren matrixartig aufgebaut.
Die Hämsynthese verläuft in mehreren Stufen unter dem Einfluss verschiedener Enzyme:
1. Die Bildung von Delta-Aminolävulinsäure erfolgt zuerst. Diese Reaktion erfolgt als Ergebnis der Kondensation von Succinyl-CoA und Glycin in Mitochondrien unter der Wirkung des Enzyms Aminolävulinat-Synthetase.
2. Die nächste Reaktion findet im Zytoplasma statt. Porphobilinogen entsteht durch die Kondensationsreaktion zweier Moleküle von Delta-Aminolävulinsäuren.
(3) Dann wird als Ergebnis mehrstufiger Reaktionen Protoporphyrin 1X aus vier Monopyrrolmolekülen des Porphobilinogens, der direkten Vorstufe von Häm, gebildet.
4. Protoporphyrin IX bindet ein Eisenmolekül (die Reaktion erfolgt unter dem Einfluss des Enzyms Hemsynthetase oder Ferrochelatase) und es entsteht Häm, das dann zur Biosynthese aller Hämoproteine verwendet wird. Beide Enzyme, die an der PBH-Synthese beteiligt sind, werden reguliert, sie werden durch Häm und Hb gehemmt. Daher wird Häm nicht im Überschuss oder Mangel gebildet. Auch der Proteinteil von Hb wird streng in einer bestimmten Menge gebildet, da seine Synthese nur in Gegenwart eines Themas erfolgen kann und die resultierenden Polypeptidketten sofort mit dem Häm kombiniert werden. Bei einer niedrigen Hämkonzentration verlangsamt sich auch die Bildung von Hämoglobin, wenn seine Synthese gestört ist.
Jede der gebildeten Polypeptidketten des Globins wird an das Code-Häm angehängt, wodurch ein monomeres Hämoglobian gebildet wird. 4 solcher Zahlen bilden, wenn sie kombiniert werden, Hämoglobin.
Die Hauptfunktion von Hämoglobin besteht darin, Sauerstoff von der Lunge in die Gewebe zu transportieren und Kohlendioxid aus den Geweben in die Lunge zu übertragen, wodurch der pH-Wert des Blutes erhalten bleibt. Hämoglobin erfüllt seine Funktionen nur als Teil eines Erythrozyten. Die Lebensdauer eines Erythrozyten beträgt 110-120 Tage. Dann wird das rote Blutkörperchen hämolysiert
3. Aufschlüsselung von Hämoglobin. Umwandlung von Bilirubin im Magen-Darm-Trakt. Freies und gebundenes Bilirubin. Eigenschaften .
Bei der Hämolyse von Erythrozyten gelangt Hämoglobin in den Blutkreislauf und verbindet sich mit dem Protein Haptoglobin, in Form eines Hämoglobin-Haptoglobin-Komplexes (Hp-Hb) wird es zu den Zellen des Makrophagen-Monozyten-Systems (MMC) transportiert: das sind Kupffer-Zellen der Leber, Zellen der Lymphknoten, Milz, Peyer-Darm.
Der Prozess beginnt mit der oxidativen Spaltung der Methinbrücke zwischen dem ersten und zweiten Pyrrolring und es entsteht Verdoglobin. Anschließend werden aus Verdoglobin Globin und Eisen abgespalten und Biliverdin (grün), eine Substanz mit linearer Struktur, gebildet. Eisen verbindet sich mit β-Globulinen und wird in Form von Transferrin an Leber und Milz abgegeben, wo es in Form von Ferritin abgelagert wird. Globin wird wie alle einfachen Proteine in Aminosäuren zerlegt.
Biliverdin wird durch NADPH 2 zu unkonjugiert reduziert,
freies Bilirubin, das in Wasser unlöslich und eine giftige Verbindung ist. Freies Bilirubin verlässt die MMS-Zellen, bindet an
Albumin und dringt in die Hepatozyten ein. Im Blut wird es als indirekt bezeichnet, weil es nicht sofort mit dem Ehrlich-Reagenz reagiert, sondern nach Zugabe eines Koffein-Reagenzes oder Alkohols zum Blutserum, um das Protein auszufällen.
In den Kupffer-Zellen der Leber beginnt auch der Abbau von Hämoglobin mit
Bildung von Verdoglobin, dann Biliverdin. In der Leber wird indirektes Bilirubin in Hepatozyten durch eine Konjugationsreaktion unschädlich gemacht und verbindet sich mit einem oder zwei Glucuronsäuremolekülen zu Bilirubinmono- oder -diglucuronid. Ein solches Bilirubin wird als konjugiert bezeichnet und
verbunden und direkt. Dieses Bilirubin löst sich gut in Wasser und hat keine toxischen Eigenschaften. Biliverdin und direktes Bilirubin werden in der Gallenblase gesammelt, wodurch die Galle eine olivfarbene Farbe erhält und daher als Gallenpigmente bezeichnet wird. Galle gelangt in den Dünndarm, aber im Gallengang wird direktes Bilirubin unter Verlust von Glucuronsäure wieder in indirektes Bilirubin umgewandelt. Biliverdin durchdringt den gesamten Darm ohne seine chemische Struktur zu verändern und wird mit dem Kot ausgeschieden, wodurch er grünlich gefärbt wird, d.h. es ist das Pigment im Kot. Und indirektes Bilirubin wird im Darm zu Mesobilinogen (Urobilinogen) reduziert, von dem ein Teil in die Pfortader aufgenommen und in die Leber zurückgeführt wird, wo es zu farblosen Mono- und Dipyrrolen zerfällt. Letztere werden zusammen mit dem Urin über die Nieren ausgeschieden.
Der größte Teil des Mesobilinogens gelangt in den Dickdarm, wo unter
der Einfluss von Enzymen von Mikroorganismen wird auf Stercobilinogen wiederhergestellt. Ein Teil des Stercobilinogens, das über die Hämorrhoidalvenen in den Blutkreislauf aufgenommen wird, gelangt in die Nieren. Im Urin wird unter dem Einfluss von Licht und Luft Stercobilinogen zu Stercobilin oxidiert, wodurch der Urin eine gelbe Farbe erhält, d.h. ist ein Urinpigment. Der Rest des Stercobilinogens wird im Dickdarm im Licht zu Stercobilin oxidiert und ist zusammen mit Biliverdin ein Farbstoff im Stuhl, der ihm eine bräunlich-grüne Farbe verleiht.
Säuglinge haben keine Fäulnisbakterien in ihrem Darm, also
Bilirubin wird nicht in Stercobilinogen umgewandelt und als solches ausgeschieden. Dementsprechend ist die Farbe des Stuhls bei Kindern auf Biliverdin und Bilirubin (gelb-grün) zurückzuführen.
Bei Kindern wird embryonales Hämoglobin in den ersten drei Monaten der Embryonalperiode gebildet. Es wird dann in fötales (Hämoglobin F) umgewandelt, das bis zur Geburt des Babys dominiert. Nach der Geburt, während des ersten Lebensmonats, wird das fetale Hämoglobin allmählich durch das erwachsene Hämoglobin (Hämoglobin A) ersetzt, das sich in der Zusammensetzung der Polypeptidketten unterscheidet. Fetales und fetales Hämoglobin haben im Vergleich zu adultem Hämoglobin eine höhere Affinität zu Sauerstoff.
Jede Pathologie im Eisenstoffwechsel wird von der Entwicklung begleitet Anämie – ein schmerzhafter Zustand, der entweder durch eine Abnahme der Anzahl roter Blutkörperchen oder eine Abnahme des Hämoglobinbeitrags gekennzeichnet ist. Dies ist eine der häufigsten Beschwerden. Es tritt aus verschiedenen Gründen auf:
a) aufgrund eines Eisenmangels in der Ernährung (bei Vegetariern, während des Fastens, bei verschiedenen Diäten zur Gewichtsreduktion, bei schwangeren Frauen, während der Stillzeit, bei schnell wachsenden Jugendlichen);
b) durch gestörte Resorption im Magen-Darm-Trakt (mit Hyposekretion von Salzsäure, Proteasen, nach subtotaler Gastrektomie, mit Verschiebungen im Nährstoffhaushalt - Mangel an Ascorbat, Succinat, Phytinsäureüberschuss, Ballaststoffen, mit Schädigung der Darmschleimhaut in peptische Ulkuskrankheit, Zwerchfellhernie , Colitis ulcerosa, nach Behandlung mit Salicylaten, Steroiden, mit Helminthiasis, insbesondere mit Läsionen mit Peitschenwurm, Hakenwurm);
c) aufgrund unzureichender Eisenspeicher;
d) aufgrund von Veränderungen in einzelnen Verknüpfungen des Stoffwechsels dieses Übergangsmetalls (mit Hemmung der Aktivität von Häm-Syntheseenzymen);
e) nach einer erhöhten Freisetzung von Ionen dieses Metalls aus dem Körper (bei akutem und chronischem Blutverlust, nach starker Menstruation - Polymenorrhoe, mit Hämorrhoiden, verschiedenen Geschwüren im Magen, Darm, nach wiederholten Episoden von Hämoptyse).
Die wichtigsten klinischen Anzeichen einer Anämie: Schwäche, Herzklopfen, Müdigkeit, Zerstreutheit, Blässe, Kurzatmigkeit.
Je nach Erhaltungsgrad der Eisenmenge im Körper werden Fe-Mangel, Fe-Mangel, Fe-Überschuss Anämien sezerniert. Etwa 98 - 99% aller Fälle solcher Erkrankungen treten bei der ersten Option auf. Andere basieren auf Anomalien bei der Verwendung von Eisen bei der Hämsynthese. Die in dieser Verbindung nicht enthaltenen Übergangsmetallionen beginnen sich in der Form abzulagern Hämosiderin (erblich und erworben Hämochromatose ) in Organen und Geweben (Leber, Bauchspeicheldrüse, Myokard, Gelenke, Haut) mit anschließender Unterdrückung ihrer Funktionen. Folgender Zeichendreiklang kann verfolgt werden: Leberzirrhose, Diabetes mellitus, Bronzefärbung der Haut (Bronzediabetes). Und da sich parallel Symptome einer Anämie entwickeln (aufgrund eines Mangels an Hämoglobin-Häm in Erythrozyten), ist es besonders gefährlich, Eisenpräparate als Therapeutikum zu verwenden.
Ein Beispiel für solche Anämien ist Methylmalonazidurie , die auf einer genetischen Schädigung der Arbeit des B12-haltigen Enzyms beruht - Methylmalonyl-CoA-Mutase , verantwortlich für die Isomerisierung von Methylmalonyl-Co A zu Succinyl-CoA - einem der Substrate bei der Häm-Genese.
Pathogenese Morbus Heilmeyer liegt daran, dass die Funktion des Gens, das für die Synthese von Transferrin verantwortlich ist, blockiert ist. In Abwesenheit funktioniert das System der Eisenfreisetzung im Knochenmark nicht, in dieser Hinsicht wird die Hämbildung unterdrückt, es entwickelt sich eine Anämie. Die Krankheit wird autosomal-rezessiv vererbt.
Krankheiten, die auf einer Schädigung der Hämsynthese beruhen, werden benannt Porphyrie ... Je nach Lokalisation der Erkrankungen werden erythropoetische (Störung des Porphyrinstoffwechsels im Knochenmark) und hepatische (ähnliche Veränderungen der Hepatozyten) unterschieden. Meistens ist dies genetisch bedingt, seltener wird es in der Natur erworben. Derzeit sind Blöcke aller an der Hämsynthese beteiligten Enzyme registriert.
Die Porphyrie (genauer ihre erbliche erythropoetische Form) wurde erstmals von Schultz (1874) und Gunther (1911) beschrieben. Die historischen Chroniken des Mittelalters haben jedoch Beschreibungen von Familien erhalten, deren Mitglieder Merkmale zeigten, die für schwere Formen dieses Leidens charakteristisch sind, die sich in Haut-, neurologischen und abdominalen Symptomen manifestieren (akute abdominale Symptome, epileptische Anfälle, Polyneuritis, Halluzinationen, Blindheit), sowie sowie eine ungewöhnlich hohe Freisetzung von Porphyrinen mit Urin oder Kot. Einige Anzeichen der Krankheit sind eine rote Färbung von Zähnen und Knochen, eine eigentümliche Hautfarbe, die durch Blasen, Geschwüre und Narben verändert wird; die nächtliche Lebensweise, die durch Photodermatitis verursacht wird, das spontane Leuchten einiger Gewebe und Sekrete des Patienten, die mit Anämie verbundenen Geschmacksmängel sind so lebhaft und ungewöhnlich, dass sie an Beschreibungen des Aussehens und Verhaltens von mythischen Ghulen oder Vampiren erinnern.
Seltener sind erworbene Porphyrien, deren Ursachen Vergiftungen mit Schwermetallsalzen sind, die in Wechselwirkung mit den Sulfhydrylgruppen der Aminolävulinatsynthase oder Ferrochelatase deren Aktivität bei der Hämgenese unterdrücken. Infolgedessen reichert sich Protoporphyrin in Erythrozyten an, der Eisengehalt steigt im Blutplasma, es lagert sich in Organen und Geweben ab und provoziert die Bildung von Hämosiderose.
Natürlich ist die Pathologie der Globinsynthese erblich. Es gibt zwei Hauptformen von Störungen: einzelne Aminosäureschäden in der Proteinstruktur ( Hämoglobinose ) und Unterdrückung der Produktion einer beliebigen Polypeptidkette von Globin ( Thalassämien ).
Derzeit sind mehr als 300 Arten von pathologischen Hämoglobinen beschrieben worden. Die ersten modifizierten Hämoproteine wurden mit lateinischen Buchstaben (C, D, E, M, S) benannt, aber als die Zahl der modifizierten Hb die Zahl der Buchstaben im Alphabet überstieg, wurden sie nach dem Fundort benannt (Kansas, Boston, San Yose, Hiroshima, Richmond usw.).
Die Heterogenität genetischer Schäden (Ersatz, Insertion, Frameshift, Kettenverlängerung usw.) führt zu verschiedenen Konsequenzen (die Affinität zu Sauerstoffveränderungen, die Stabilität der Erythrozyten nimmt ab, was sich in einer erhöhten Hämolyse, Zyanose äußert). Dies wird nicht nur durch die Schwere der Verschiebungen in der Nukleotidsequenz bestimmt, sondern auch durch die Art der veränderten Aminosäuren. Wenn Analoga ersetzt werden, zum Beispiel Glutamat durch Aspartat, dann manifestiert sich diese Option in keiner Weise. Praktisch gesund sind auch die Träger von Hb San Jose, bei denen in der 7. Position der Beta-Kette Glutamat durch Glycin ersetzt wird, was nur die elektrophoretische Mobilität dieses Proteins beeinflusst.
Wenn sich die Struktur der mutierten Stelle abrupt ändert, ist die Wahrscheinlichkeit hoch, schwere klinische Symptome zu entwickeln. Besonders gefährlich sind Läsionen, die in den Kontaktbereichen (die Stellen, an denen einzelne Untereinheiten zu Tetrameren binden) oder in den Taschen lokalisiert sind, in denen sich das Häm befindet. In diesen Fällen ist die Komplexierung des heterogenen Tetramers gestört, was die Lebensfähigkeit des Embryos beeinträchtigt und die Wahrscheinlichkeit einer Fehlgeburt erhöht. Ein Beispiel ist Hb Philly, bei dem Phenylalanin Tyrosin ersetzt, das mit anderen Untereinheiten in einem normalen Protein eine Wasserstoffbrücke bildet. Nach einer solchen Mutation wird die Existenz eines genetisch veränderten Tetramers unmöglich – die Mizelle zerfällt. Boston Hb ist durch die Substitution von Histidin durch Tyrosin an Position 58 der Alpha-Kette gekennzeichnet. Histidin hingegen bildet gewöhnlich eine Koordinationsbindung mit Häm-Eisen, und Tyrosin oxidiert das Ion zu Eisenphenolat; das resultierende Methämoglobin provoziert Hypoxie.
Beim Genowa-Hämoglobin wird durch den Verlust von Valin ein Fragment verdrängt, der Rest des Glutamats befindet sich in der Mizelle, die Stelle ist deformiert, was die Affinität für Sauerstoff verringert, und es entwickelt sich eine Zyanose.
Günstigere Folgen sind möglich, wenn Mutationen die Aminosäuren betreffen, die die Micellenoberfläche bilden. Ein klassisches Beispiel ist Hb S, bei dem in der 6. Position der Beta-Kette Glutamat durch Valin ersetzt wird, also eine saure Verbindung durch ein Hydrophob ersetzt wird. Und da der Schaden an der Oberfläche ist, verringert eine solche Änderung die Ladung und Löslichkeit von Hämoglobin, und seine einzelnen Moleküle kollidieren aufgrund der Bildung hydrophober Wechselwirkungen von Valinen verschiedener Micellen miteinander. Eine solche Aggregation verlängert die Hämoproteinfilamente, was die Löslichkeit weiter verringert. Dieses Phänomen beeinflusst die Form der roten Blutkörperchen: Sie nehmen die Form einer Sichel an (Sichelzelle - Sichelzelle). Daher wird die Krankheit als sichelförmige Anämie bezeichnet, oder Hämoglobinose S . Die beschädigte Erythrozytenmembran ist weniger stabil, was Hämolyse und Thrombose hervorruft. Die Patienten sind gekennzeichnet durch hämolytische Krisen mit akutem Schmerzsyndrom, Symptomen einer Leberschädigung, intensiver Gelbsucht und wahrscheinlich Steinbildung in den Gallenwegen. Die Häufigkeit des Vorhandenseins eines solchen Hämoproteins in den Vereinigten Staaten beträgt 8-9% (bei Afroamerikanern) und in einigen Regionen Griechenlands erreicht sie 40%.
Thalassämien(Thalassa - Meer) - Erbkrankheiten, die, wie oben erwähnt, auf dem Syntheseblock der gesamten Globinkette basieren. Gleichzeitig werden in erythroiden Zellen Mizellen synthetisiert, die oft homogene Tetramere sind, die beispielsweise nur aus Beta- oder Gamma-Ketten bestehen. Hämoglobin H, bestehend aus 4 Beta-Untereinheiten, kann nur Sauerstoff binden, aber nicht abgeben. Mit der Hemmung der Synthese von Alpha-Ketten ist der homozygote Embryo nicht lebensfähig, es kommt zu einer Fehlgeburt in der Frühschwangerschaft. Kind mit Beta-Thalassämie (Cooly Disease) (ein Geneseblock der Beta-Kette) wird praktisch gesund geboren (schließlich wird Hämoglobin, einschließlich Alpha- und Gamma-Ketten, für das Wachstum und die Entwicklung des Fötus benötigt). Jeden Monat treten pathologische Anzeichen auf, die durch den Grad der Hypoxie, erhöhte Lipidperoxidation, Schädigung der Erythrozytenmembranen (schwere hämolytische Anämie, Persistenz eines hohen HbF-Gehalts (bis zu 20-30%), beeinträchtigte körperliche und geistige Entwicklung, die Kind weigert sich zu brusten, das Gesicht bekommt mongoloide Züge - die Wangenknochen ragen nach vorne, der Nasenrücken wird eingedrückt, die Nase wird abgeflacht). Für ein höheres Alter sind hämolytische Krisen, ein fieberhafter Zustand charakteristisch, und es kann sich eine Herzinsuffizienz entwickeln.
Die Verbreitung dieser Krankheiten variiert je nach Region. Besonders häufig sind sie an den Ufern der Meere (Mittelmeer, Schwarz). In Transkaukasien sind bis zu 10 % der Träger solcher Gene registriert.
DISHEMOGLOBINÄMIE
Pathologische Zustände sind relativ häufig, die auf Ungleichgewichten in verschiedenen Formen von Hämoglobin beruhen. In den Erythrozyten eines gesunden Erwachsenen ist der Spiegel von Methämoglobin 2% des Gesamtinhalts nicht überschreiten. Unter dem Einfluss verschiedener Stickoxide, anorganischer Nitrate, organischer Nitroverbindungen (Amylnitrit, Nitrobenzol, Nitrophenol, Trinitrotoluol, Nitroanilin), Aminoderivate (Hydroxylamin, Phenylhydrazin, Aminophenole, p-Aminobenzoesäure, Anilin), Chlorate, Chromate, Permanganate, Pyron einige Medikamente (Nitroglycerin, Anestezin, Furadonin, Barbiturate, Aspirin usw.), Farben mit Oxidationsfähigkeit - ihre Konzentration steigt stark an ( Met-Hämoglobinämie ). Dadurch wird die Hauptfunktion des Hämoglobins gestört - der Sauerstofftransport von der Lunge in das Gewebe wird blockiert und es entwickelt sich eine Hypoxie.
Alle Methämoglobin-Bildner, die die osmotische Resistenz von Erythrozyten reduzieren, beschleunigen ihre Hämolyse. Bei der Oxidation von Hb zu Methämoglobin entstehen aktive Sauerstoffradikale, die an den Prozessen der oxidativen Schädigung von Erythrozyten teilnehmen können. LPO wird aktiviert, der Fettstoffwechsel der roten Blutkörperchenmembranen ist gestört, das Gleichgewicht im LPO - AOD-System ist verschoben. Das Hauptsymptom ist Zyanose; wenn der Methämoglobingehalt 30% überschreitet, treten Schwäche, Schwindel, Tachykardie, Kopfschmerzen auf; wenn es sich bis zu 50% ansammelt, entwickelt sich ein Herz-Kreislauf-Versagen. Das Risiko einer Nitritvergiftung steigt mit der Verwendung von Gemüse, Wurst, geschmortem Fleisch und minderwertigem Trinkwasser. Auch Fälle von hereditären Methämoglobinämien (bei Trägern von Hb Boston) wurden beschrieben (so).
Zur Gruppe der Blutgifte, die pathologische Pigmente bilden, gehören Kohlenmonoxid (Kohlenmonoxid). In den Körper eindringendes CO wird von Erythrozyten aufgenommen, interagiert mit dem Hämoglobin-Eisen und bildet eine ziemlich stabile Verbindung - Carboxyhämoglobin , dessen Wert bei Nichtrauchern 0,25 % der Gesamtmenge des basischen Erythrozytenproteins nicht überschreitet. Im Blut von Rauchern steigt seine Zahl auf 6-7%. Bei einem höheren Kohlenmonoxidpartialdruck ( Carboxyhämoglobinämie ) wird die Oxygenierung des Hämoproteins gehemmt, es entwickelt sich eine Hypoxie. Darüber hinaus hat CO die Fähigkeit, mit anderen Häm-haltigen Proteinen (Myoglobin, Cytochrome, Peroxidase, Katalase) in Kontakt zu kommen und deren Funktionen zu stören.





